CHAPTER 37 Malignant Hyperthermia
Deaths during anesthesia associated with high fever and tachycardia were described repeatedly in the first half of the 20th century (Moschcowitz, 1916; Burford, 1940). These deaths were sometimes ascribed to heat stroke and dehydration, because at that time intravenous hydration and air conditioning were not available. In 1915 and 1919, two deaths in one family occurred during chloroform anesthesia; in these cases, severe muscle spasm was observed before cardiac arrest. In 1987 a young child from this same family died during anesthesia for dental surgery. Subsequent in vitro contracture testing demonstrated susceptibility to malignant hyperthermia (MH) in several relatives in the family (Harrison and Isaacs, 1992). Even though MH is often suspected in cases such as these, in the 21st century it can still take years to complete MH diagnostic testing for a family.
Denborough was the first to investigate MH as a clinical entity (Denborough and Lovell, 1960; Denborough et al., 1962). Dr. James Villiers anesthetized a young Australian man for repair of a broken leg; the man was afraid to undergo general anesthesia because many of his relatives had died during ether anesthesia. Although during halothane anesthesia the patient became febrile, tachycardic, cyanotic, and hypotensive, he was aggressively cooled in the recovery room and he survived. Villiers asked his medical colleagues to investigate this patient and family. They found a pattern of anesthetic deaths consistent with an autosomal dominant trait. In 2009, despite many investigations, the genetic change associated with abnormal muscle calcium homeostasis in this family was still not identified (Ball, 2007).
An episode of MH is recognized by documentation of an increasing metabolic rate after exposure to a triggering agent such as a potent inhalation anesthetic. Rhabdomyolysis may also be observed. Susceptibility to MH is rarely associated with clinically evident hypermetabolism in the absence of drugs used during anesthesia (Gronert et al., 1980; Tobin et al., 2001). Halothane and succinylcholine, two drugs formerly used often in pediatric anesthesia, are two of the most potent triggers of MH. A review of 25 years of halothane anesthetics in one pediatric hospital found the incidence of acute MH episodes to range from 1:20,000 to 1:40,000 anesthesia procedures (Warner et al., 1984). Because choices of anesthetic and neuromuscular blocker influence the development of this potentially lethal syndrome, MH is a disease of particular concern to the pediatric anesthesiologist who chooses to administer inhalation anesthetics or succinylcholine. Indeed, in 2008 administration of succinylcholine during potent inhalation anesthesia precipitated cardiac arrest and death from MH (North American Malignant Hyperthermia Registry, 2008). The purpose of this chapter is to review the clinical manifestations, management, and underlying pathophysiology of MH.
Muscle physiology
Normal Muscle Physiology
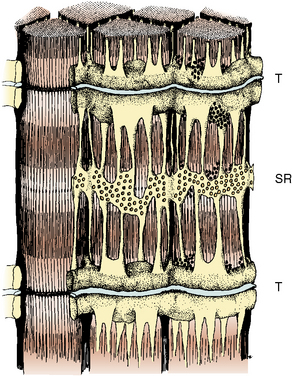
The propagation of an action potential down a motor nerve fiber produces depolarization of that fiber and release of acetylcholine (ACh) at the neuromuscular junction. When two molecules of ACh interact with the ACh receptor, the channel opens, allowing sodium to enter the cell and potassium to flow out. This initiates a wave of depolarization along the muscle cell membrane or sarcolemma. The propagated depolarization wave spreads internally via transverse tubules that abut onto the sarcoplasmic reticulum (SR) (Fig. 37-1). The SR is an internal cellular structure and a major storage site for calcium ions. The wall of the transverse tubule contains the dihydropyridine receptor, a voltage-dependent calcium channel that interacts with the calcium-sensitive ryanodine receptor (RYR), the footplate between the transverse tubule and the SR. Within a voltage window, calcium is released from the SR (Melzer and Dietze, 2001) into the sarcoplasm through RYR type 1 (RYR1). Increased calcium in the sarcoplasm also induces further calcium release from the SR.
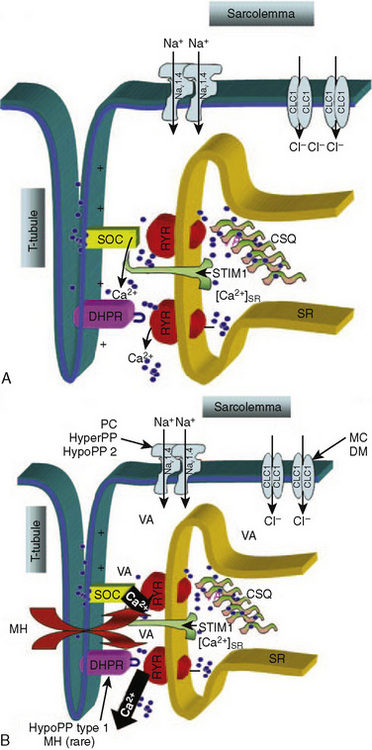
Furthermore, when calcium has been depleted from the SR, a process known as store-operated calcium entry (SOCE) allows calcium entry into the sarcoplasm from the extracellular milieu (Fig. 37-2) (Zhao et al., 2006). Excitation-coupled calcium entry (ECCE) refers to the process by which extracellular calcium enters the sarcoplasm through the plasma membrane after depolarization (Cherednichenko et al., 2004; Lyfenko and Dirksen, 2008). The RYR has influence on both SOCE and ECCE.
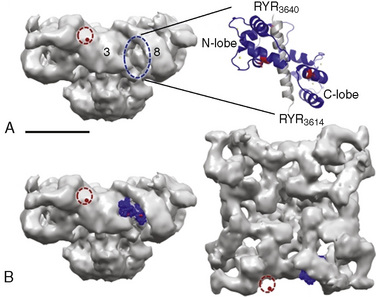
Type 1 RYRs are found in all skeletal muscle, smooth muscle, neurons, and B lymphocytes. Type 2 RYRs are found in cardiac muscle, brain, and some hematopoietic cells. Type 3 RYRs are found in skeletal and smooth muscle and to a lesser extent in the brain. The function of RYR1 is modified by other proteins and by the redox state of the cell (Fig. 37-3) (Aracena-Parks et al., 2006; Goonasekera et al., 2007; Durham et al., 2008; Cornea et al., 2009; Protasi et al., 2009).
Pathophysiology of Malignant Hyperthermia
In classic MH, mutation of the RYR1 gene produces enhanced activation of RYR1 by intraluminal calcium and reduced threshold for spontaneous calcium release during store calcium overload (store-overload–induced calcium release) (Jiang et al., 2008). In pigs, a specific single-point mutation in the RYR1 exists in all major breeds that are susceptible to malignant hyperthermia (Fuji et al., 1991). This point mutation is seen in a minority of human families who are susceptible to MH; however RYR1 on chromosome 19q13.1 has been linked to MH susceptibility in the majority of families studied. Of the four other loci proposed as linked to MH susceptibility, only the CACNA1S on chromosome 1q13.1, coding for the α1 subunit of the dihydropyridine receptor, has been linked to MH in more than two human families (Carpenter et al., 2009). The heterogeneous nature of human MH may be the result of a number of different mutations in RYR1 or to abnormalities in other proteins such as the dihydropyridine receptor (Gallant and Lentz, 1992). In humans who have MH susceptibility, RYR1 compound heterozygotes have been observed in several countries. This supports the claim that the incidence of a MH-causative genotype could be as great as 1 in 3000 people (Monnier et al., 2002; Ibarra et al., 2006).
The metabolic dysfunction of MH is caused by increased intracellular ionized calcium (Mickelson and Louis, 1996). A direct result of increased intracellular calcium is an increased need for intracellular ATP to drive pumps that transfer calcium into the SR, across the sarcolemma into the extracellular fluid, or into mitochondria. Increased demands for ATP lead to the clinically detectable manifestations of MH (e.g., an increase in metabolism). The elevation of intracellular calcium observed in MH muscle is decreased by dantrolene (Lopez et al., 1985; Mickelson and Louis, 1996; Cherednichenko et al., 2008).
Motor nerve activity and neuromuscular transmission are normal in patients who have MH susceptibility (Gronert, 1980). However, susceptible porcine muscle has a lower mechanical threshold than does normal muscle, and compared with normal muscle, less depolarization is required to initiate contraction (Moulds and Denborough, 1974; Okumura et al., 1979; Gronert, 1980). Calcium release from the SR occurs at more negative potentials in the presence of RYR1 MH mutations, and calcium-induced calcium release from the SR is abnormal in subjects susceptible to MH. The abnormal RYR1 is resistant to the inhibitory effects of both calcium and magnesium (Mickelson and Louis, 1996).
During an episode of MH in pigs, lactate release from muscle increases before mixed venous oxygen tension decreases (Gronert and Theye, 1976a). This sequence is consistent with intracellular ATP depletion. Magnetic resonance imaging (MRI) techniques showed an increased ratio of inorganic phosphate to phosphocreatine in vivo in human muscle susceptible to MH. This ratio is an indicator of the energy state of the muscle (Chance et al., 1985a, 1985b). An increased ratio suggests either impaired synthesis of ATP or increased breakdown of ATP.
Mechanism of Action of Dantrolene
Since dantrolene was introduced in the 1970s, its use has remarkably improved the treatment and survival of patients with MH. It has been claimed that dantrolene inhibits the release of calcium from the SR by limiting the activation of the calcium-dependent RYR1, but more recently it was shown that dantrolene blocks ECCE across the plasma membrane (Fruen et al., 1997; Cherednichenko et al., 2008). Dantrolene does not act at the neuromuscular junction and has no effect on the passive or active electrical properties of the surface and tubular membranes of skeletal muscle fibers. Patients who have been given dantrolene have normal neuromuscular transmission and depressed force of muscle contraction. The protective effects of dantrolene against MH require significant depression of the force of contraction (Flewellen et al., 1983). Dantrolene can produce clinically significant weakness and decreased metabolism in normal muscle as well as in MH-susceptible muscle (Flewellen et al., 1983). It can reduce elevated creatine kinase (CK) in diseased muscle, independent of the presence of RYR1 mutations (Bertorini et al., 1991; Kamper and Rodemann, 1992).
The dose-response relationship of dantrolene in children has not been reported. Available data suggest that the half-life of dantrolene in children is somewhat shorter than that in adults: 7.3 to 9.8 hours and 12.1 hours, respectively (Fig. 37-4) (Lietman et al., 1974; Flewellen et al., 1983; Lerman et al., 1989). In adults, a cumulative dose of 2.2 to 2.5 mg/kg of dantrolene administered intravenously over 125 minutes produced a steady plasma concentration of dantrolene for longer than 5 hours (Flewellen et al., 1983). Orally administered dantrolene, a total of 5 mg/kg in 3 or 4 divided doses administered every 6 hours to adults with MH susceptibility, has also been shown to produce protective plasma concentrations of dantrolene for at least 6 hours after induction of anesthesia (Allen et al., 1988). In children, intravenous administration of 2.4 mg/kg of dantrolene infused over 10 minutes produced stable blood levels of about 3.5 mcg/mL for 4 hours, after which a slow decline in plasma concentration occurred (Lerman et al., 1989). Hence, it may be reasonable to repeat 1 mg/kg of dantrolene every 5 to 7 hours for prophylaxis. Alternatively, pharmacokinetic modeling with parameters derived in adults recommends a continuous infusion of dantrolene (Podranski et al., 2005). It is likely that when plasma concentrations of dantrolene are sufficient to inhibit an episode of MH, the patient experiences weakness and possibly disequilibrium.
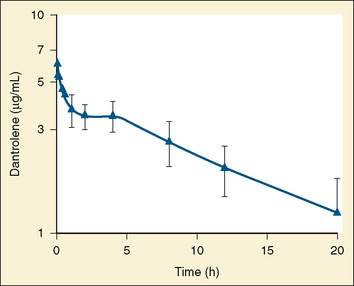
FIGURE 37-4 Plasma decay of dantrolene in children.
(From Lerman J et al: Pharmacokinetics of intravenous dantrolene in children, Anesthesiology 70:625, 1989.)
A dynamometer could be used to assess grip strength objectively, but this requires the patient’s cooperation. In a study of adults, the dose of dantrolene that produced maximal depression of grip strength and evoked force of thumb contraction had no significant effect on vital capacity (Flewellen et al., 1983). Similar studies have not been performed in children. Clinical experience suggests that less than 2 mg/kg of dantrolene administered intravenously to a child preoperatively can be associated with significant hypotonia in the postoperative period (Brandom and Carroll, unpublished observations). Weakness induced by dantrolene could compromise the ability to swallow and even necessitate artificial protection of the airway and mechanical ventilation, although this has never been reported in the literature (Flewellen et al., 1983). Intravenous dantrolene should be administered in settings where support of airway and ventilation can be easily provided.
Clinical presentation
The initial clinical signs of an impending episode of MH are nonspecific. The most commonly reported initial signs are hypercarbia, sinus tachycardia, and masseter spasm or jaw rigidity (Larach et al., 2010). Arrhythmias and tachypnea may be present (Box 37-1). Increased or rapidly increasing temperature is often an early sign of MH, and metabolic acidosis is often not observed before treatment of MH (Larach et al., 2010).
Box 37-1 Steps in Identifying and Treating Malignant Hyperthermia
Clinical events during MH
Rigidity of the extremities and the entire body are signs of fulminant MH. When MH is fulminant, there is severe metabolic acidosis (base deficit often greater than 8 mEq/L), respiratory acidosis (arterial carbon dioxide tension [Paco2] of greater than 60 mm Hg), tachycardia with arrhythmias, a rapid increase in body temperature to 39.5° C or greater, hyperkalemia, myoglobinuria, and often, but not always, a marked increase in serum CK (Gronert, 1980; Newmark et al., 2007). However, because of modern comprehensive monitoring and increased awareness of MH, fulminant MH is rarely observed. The clinical diagnosis of MH is often considered before metabolism and temperature reach these extremes (Karan et al., 1994). The patient’s medical history and clinical course usually, but not always, help to differentiate fulminant MH from other metabolic crises such as porphyria, thyroid storm, untreated pheochromocytoma, and Ecstasy exposure (Allen and Rosenberg, 1990). Some other conditions that produce signs similar to MH are sepsis, allergic drug reactions, intracranial trauma, and hypoxic encephalitis.
Ideally, MH should be recognized and treated before it becomes fulminant. Treatment is more likely to be successful the earlier it is started, but because the signs of MH are not highly specific it is difficult for anesthesia practitioners to distinguish between early MH and many other complications. If there are only mild symptoms or signs suggestive of MH (e.g., moderate increases in heart rate, blood pressure, and temperature along with a slight metabolic or respiratory acidosis), yet evidence is later obtained that the patient is indeed susceptible to MH, this constellation of clinical findings could be called abortive MH. In such cases, masseter spasm may or may not occur (Ellis et al., 1990). There may be a moderate increase in CK and serum myoglobin. When considering the implications of these tests, note that myoglobin appears in the plasma within minutes of muscle injury by succinylcholine. However, CK continues to increase for 8 to 20 hours after a transient injury, even in patients who are not susceptible to MH (Florence et al., 1985). The incidence of abortive MH is as high as 1:4200 anesthesias when succinylcholine is used in combination with potent inhalation anesthetics (Ording, 1985). However, in the first decade of the 21st century, the incidence of MH cases identified by hospital discharge diagnosis codes was estimated to be about 1:100,000 anesthesias (Brady et al., 2009).
The diagnosis of MH may be mistakenly applied to patients suffering hyperkalemic cardiac arrest or sudden exacerbation of chronic rhabdomyolysis, which has occurred in pediatric patients after the administration of succinylcholine (Kovarik and Morray, 1995; Larach et al., 1997; Piotrowski and Fendler, 2007). In this condition, the sudden severe increase in plasma potassium can be fatal, but metabolic abnormalities are secondary to cardiac failure and not increased metabolism in skeletal muscle, as in MH (Delphin et al., 1987; Rosenberg and Gronert, 1992; Tang et al., 1992; Larach et al., 2001).
In a group of 48 children, 17 of whom later proved to be susceptible to MH by muscle biopsy, two or more adverse signs or abnormal laboratory findings were present in all patients with positive in vitro contracture tests (Larach et al., 1987). However, similar adverse events occurred in 83% of the children who had negative muscle-biopsy findings. Generalized muscle rigidity was the single factor significantly associated with positive biopsy findings for MH. However, generalized muscle rigidity is not an absolute predictor of MH susceptibility. Three of 24 patients, who were referred for muscle-contracture testing to evaluate MH susceptibility and had negative contracture test results, had experienced generalized muscle rigidity during induction of anesthesia. Signs consistent with abortive MH, such as tachycardia, premature ventricular contractions, elevated end-tidal carbon dioxide, and increase in tension of the masseter muscle may be observed in the normal pediatric patient who is given halothane or sevoflurane and succinylcholine (Van der Spek et al., 1987; 1988; Lanier et al., 1990). Larach and others (1987) could not identify the patient with MH susceptibility on the basis of these signs (Hackl et al., 1990). Therefore, when MH is suspected clinically, after treating the patient arrangements should be made for confirmatory testing (see related sections in this chapter).
If events during the induction of anesthesia require explanation beyond light anesthesia, transient rigidity and tachycardia during inhalation of sevoflurane, or hypoventilation, further investigation to rule out the diagnosis of MH must be undertaken immediately, before surgery begins. The anesthesiologist must document the presence of increased metabolic rate, rather than the decreased metabolic rate that usually follows induction of anesthesia. Venous or arterial blood should be obtained for measurement of partial pressure of carbon dioxide [Pco2], lactate, potassium, and CK. Mixed venous blood is most likely to show significant alterations in Pco2, but it may not be readily available (Gronert and Theye, 1976a, 1976b). During anesthesia there is increased arterial-to-venous shunting through the skin. Despite this fact, blood from a large peripheral vein, femoral or antecubital, may demonstrate increasing Pco2 and worsening base deficit before these changes are found in arterial blood. Increasing end-tidal carbon dioxide concentrations, particularly with increased minute ventilation, supports the presumptive diagnosis of MH. Evidence of muscle injury, such as the presence of myoglobin in the serum and urine and elevated CK and other enzymes in the blood, may not be observed if MH is treated very quickly. With anesthesia techniques common in the 21st century, MH may not be recognized until the second or third hour of anesthesia. MH may first be recognized in the postoperative recovery room. Fatal MH can occur without exposure to anesthetics (Brown et al., 2000). Once a hypermetabolic state is recognized, appropriate actions must be taken without delay, as described later.
Masseter spasm
Masseter spasm (also termed masseter muscle rigidity or trismus) is a marked increase in tension of the masseter that prevents opening of the mouth when succinylcholine has produced neuromuscular blockade. Masseter spasm may be an early sign of MH. However, succinylcholine can produce increased tension in normal muscle at the same time that it produces block of neuromuscular transmission (van der Spek et al., 1987, 1988). Many anesthesiologists believe that only if the jaw cannot be forced open should this phenomenon be called masseter spasm.
Hannallah and Kaplan (1994) distinguish between masseter muscle rigidity and trismus. In masseter muscle rigidity, the mouth cannot be fully opened even with firm pressure on the incisors, but the mouth can be opened far enough to permit intubation of the trachea. In trismus, the mouth cannot be opened enough to permit intubation of the trachea. Using these definitions, Hannallah and Kaplan (1994) noted rigidity in 0.2% of 500 children anesthetized with halothane and administered succinylcholine, but none of these 500 patients experienced trismus. It may be that the several-fold greater incidence of masseter spasm noted in the 1980s included cases of incomplete jaw relaxation (i.e., the mouth opens fully with firm manual separation of the teeth), which was observed in 4.4% of these 500 pediatric patients. The 22 patients with incomplete jaw relaxation in this study continued to receive halothane anesthesia with no apparent complications. Masseter spasm has been touted as a specific early warning sign of MH. Undoubtedly, deaths from MH have occurred during anesthetic procedures in which masseter spasm was observed and in cases in which it was not observed. It may be that when masseter spasm is accompanied by rigidity of the entire body, MH is likely to occur. However, transient increase in jaw stiffness, or resting tension of jaw muscles, is a normal response to succinylcholine (van der Spek et al., 1987, 1988; Plumley et al., 1990). Increase in masseter muscle tension occurs in normal mammals after the administration of succinylcholine with prior administration of epinephrine (Pryn and van der Spek, 1990). There is a greater increase in jaw tension after administration of succinylcholine during halothane anesthesia than in the presence of barbiturates. Jaw tension is also increased in animals that are febrile as opposed to those that are normothermic (Storella et al., 1993). Temporomandibular joint abnormalities may confuse the diagnosis of masseter spasm by interfering with jaw opening.
Although masseter spasm usually occurs after anesthesia induction with halothane and the administration of succinylcholine, it may occur with other anesthetic agents (Larach et al., 1987; Marohn and Nagia, 1992; Takamatsu et al., 1996). Masseter spasm occurs despite abolition of evoked muscle function in the extremities. Tachycardia or other nonspecific arrhythmias may accompany masseter spasm. MH may follow masseter spasm immediately; in the continued presence of anesthetic trigger agents, however, a period of 10 or more minutes often intervenes between masseter spasm and the clinical presentation of MH when the clinical presentation of MH is defined as arterial Pco2 greater than or equal to 50 mm Hg, pH less than 7.25, and base deficit more negative than 8 mEq/L (O’Flynn et al., 1994).
Anesthesia with halothane has been continued after isolated masseter spasm with no increased metabolism or cardiovascular instability. Littleford and others (1991) reported on 57 such children, of whom 33% experienced transient arrhythmias intraoperatively. Most of these children also had some degree of hypercarbia or metabolic acidosis. CK levels measured 18 to 24 hours postoperatively were elevated in all but one of these children, and CK levels greater than 20,000 units/L were observed in many. However, there were 11 children who experienced generalized rigidity in combination with masseter muscle spasm. Anesthesia was aborted for four of these children and continued without inhalation agents in three of the children. None of these children developed fulminant MH in the perioperative period. The remaining four patients who developed generalized rigidity received dantrolene. Kaplan and Rushing (1992) documented a case of masseter spasm in which clinical abnormalities prompted administration of dantrolene, and postoperative CK was 40,000 IU. Nine years later, this healthy adolescent had completed extensive evaluation for neuromuscular disorders, including in vitro testing for MH with the caffeine-halothane contracture test (CHCT). The patient and family remained without signs, symptoms, or diagnosis of any myopathy. If this patient had been labeled as susceptible to MH, it would have been a misdiagnosis. Total body rigidity accompanying masseter muscle rigidity does not absolutely guarantee that the patient has MH (Larach et al., 1987). Anesthetic depth may have been misjudged; however, if rigidity does not abate when deep neuromuscular block was produced with nondepolarizing blockers, muscle must be abnormal. Either fulminant MH is occurring or the patient may have occult myotonia (Neuman and Kopman, 1993).
Inhalation anesthesia without succinylcholine was associated with fewer episodes of both fulminant (2 vs. 8) and abortive (17 vs. 110) MH than was succinylcholine with potent inhalation anesthetics in a Danish population (Ording, 1985). Thus, avoiding succinylcholine administration to pediatric patients anesthetized with inhalation anesthetics not only avoids the diagnostic uncertainties associated with masseter spasm but also produces fewer episodes of MH and other adverse events (Delphin et al., 1987; Rosenberg and Gronert, 1992). Pediatric anesthesiologists may choose to administer succinylcholine only when definite, strong indications for this drug have been identified.
Management of Masseter Spasm
There is no agreement among experienced clinicians concerning the preferred management of patients with incomplete relaxation of the masseter after the administration of succinylcholine (Kaplan et al., 1993). If jaw stiffness is mild, so that the mouth can be opened with increased effort, there is no rigidity in the rest of the body, and cardiovascular function is stable, anesthesia may be continued with careful documentation of capnography and core temperature. Fluid deficits should be replaced completely so that urine output is greater than 3 mL/kg per hour. Urine should be obtained in the early postoperative period to check for the presence of myoglobin. Blood should be obtained for measurement of electrolytes and CK. It is not necessary to terminate anesthetic administration unless signs of increasing metabolic rate occur. If jaw stiffness is so great that the mouth cannot be opened, there are several reasons to terminate elective anesthetic administration, not the least of which may be the need to clear the upper airway. If trismus and extensor rigidity are present, biochemical signs of MH should be sought and treatment of MH initiated. Venous blood should be obtained for gas analysis and measurement of electrolytes, myoglobin, and CK. If rigidity persists in the presence of neuromuscular blockade either the patient has MH or a form of myotonia.
After an episode of masseter spasm, myalgia and occasionally weakness may be present for several days or longer. In patients who were not susceptible to MH and undergoing ophthalmic surgery with halothane anesthesia who received succinylcholine intraoperatively, an increase in CK level was noted 24 hours after surgery. The highest postanesthetic CK level in these otherwise healthy patients was 40 times that of normal (Inness and Stromme, 1973). Myoglobin appears quickly in the plasma after halothane anesthesia and succinylcholine administration, even in children who had no masseter spasm (Plotz and Braun, 1982). If radioimmunoassay is used to measure serum myoglobin concentrations, increases in myoglobin can be measured within the first hour after succinylcholine administration in normal children anesthetized with isoflurane or halothane.
Postoperative renal failure has occurred in patients who had myoglobinuria after administration of succinylcholine during anesthesia. In any situation in which injury to muscle may occur, it is important to document that myoglobinuria is not present. If increased muscle stiffness is noted after administration of succinylcholine or the if child complains of muscle pain postoperatively, urine should be obtained. If there is no blood in the urine as assessed by orthotolidine (Hematest), then there is no myoglobin present (Bosch et al., 2009). If the response to blood is positive on the dipstick, urine should be examined for the presence of red blood cells, and free hemoglobin and myoglobin should be measured. If myoglobin is present, the patient should remain in the hospital. The patient should be observed for signs of MH, evaluated for the presence of occult muscle disease, and well hydrated. Alkaline urine may reduce the risk of renal tubular injury from myoglobin (Elsayed and Reilly, 2010).
Treatment of an acute episode of malignant hyperthermia
When the diagnosis of MH is strongly suspected, the most important step to take is to administer dantrolene. The other steps in management are to discontinue the triggering anesthetic agents immediately, increase minute ventilation several-fold with 100% oxygen at the highest possible flow rate, check core temperature, and alert the surgeon that the procedure must be concluded promptly. Other anesthesiologists, paramedical personnel, or both should be called in at once for assistance. If core temperature is higher than 39° C, cooling measures should be applied until temperature is below 38° C (see www.mhaus.org).
Dantrolene must be diluted with sterile, preservative-free, distilled water that should be stored in large quantities with the drug (Table 37-1). It is important to store sterile water in clearly labeled containers of a different size from those used for routine intravenous solutions and to keep a mixing system nearby. New formulations of dantrolene may go into solution faster than did dantrolene manufactured before 2008. The initial intravenous dantrolene dose should be 2.5 mg/kg, although more than 10 mg/kg may be needed to control the episode (Larach et al., 2010). Repeated dosing should be guided by clinical and laboratory signs. A flow sheet including minute ventilation, end-tidal carbon dioxide concentration, heart rate and rhythm, arterial blood pressure, central venous pressure, core temperature, and urine output, along with arterial and venous blood gas tensions, serum electrolytes, and glucose and total fluid intake, provides a useful guide for continued therapeutic interventions. Dantrolene must be administered until respiratory and metabolic acidosis, tachycardia and stiffness have resolved. The usual upper limit of 10 mg/kg may be exceeded as necessary. The most common side effects of this drug are muscle weakness and phlebitis.
TABLE 37-1 Drugs and Dosages Used to Treat an Acute Episode of Malignant Hyperthermia
| Dantrolene* | 2.5-10 mg/kg or more (Sterile water must be available to dilute dantrolene.) |
| Sodium bicarbonate | 2 mmol/kg as needed |
| Iced normal saline solution | As needed (10-12 L for 50-kg patient) |
| Mannitol | 300 mg/kg (Note that there is 150 mg of mannitol per milligram of dantrolene in the vial.) |
| Furosemide | 0.5-1 mg/kg |
| Insulin (regular) | 10 units regular insulin in 50 mL of 50% dextrose titrated to produce normokalemia |
| Lidocaine | 1 mg/kg |
* Dantrolene administration should be repeated until physical and chemical signs have returned to normal. When this degree of physiologic stability has been obtained, dantrolene (1 mg/kg or more) should be repeated approximately every 6 hours until all signs of MH, including muscle stiffness, have abated and creatine kinase has decreased consistently. If the patient is metabolically stable, vital signs are normal, and muscle weakness is present, the interval between dantrolene doses can be increased to 8 hours.
The anesthesia machine need not always be switched to a standby unit that has been kept free of inhalation anesthetics. However, because of both the increased internal gas volume of and the increased anesthetic solubility in modern anesthesia work stations, more time may be required to eliminate traces of anesthetic. For example, over 100 minutes of 10 L/min fresh gas flow (10 L/min FGF) was needed to reduce residual inhalation anesthetic to 5 ppm in the Drager Fabius anesthesia machine, whereas after 20 minutes residual sevoflurane was less than 5 ppm in the Drager Narcomed GS machine (Gunter et al., 2008; Whitty et al., 2009). Isoflurane washout can require more than 45 minutes of 10 L/min FGF in the Drager Primus machine, more than 50 minutes in the Datex-Ohmeda machine, and more than 75 minutes in the Aestiva machine (Crawford et al., 2007; Birgenheier et al., 2009). If the carbon dioxide absorber and circuit tubing are not changed, 30 minutes of 10 L/min FGF is needed to reach 2 ppm isoflurane in the Datex-Ohmeda work station (Schonell et al., 2003). In summary, replacement of all exposed elements, including the ventilator diaphragm and breathing system with autoclaved or new parts, provides the fastest preparation of the anesthetic work station without the addition of charcoal filters. Continuation of 10 L/min FGF for the duration of the anesthesia has been recommended to avoid increasing concentrations of agent (Crawford et al., 2007; Whitty et al., 2009). If MH occurs after the first hour of exposure to an inhalation anesthetic, there is a substantial volume of the anesthetic trigger agent in the patient’s body. It is likely that placement of a charcoal filter on the inspiratory limb of the anesthesia circuit and administration of 10 L/min FGF could greatly speed elimination of potent anesthetic from the breathing circuit (Gunter et al., 2008; Birgenheier et al., 2009.)
Procedures to cool the body should be instituted quickly. The goal is to reduce muscle metabolism and avoid exposure to a critical core temperature of greater than 40° C (Bouchama and Knochel, 2002). Furthermore, a temperature that is higher than normal may accelerate an MH episode in humans, as has been observed in animals (Chelu et al., 2006; Dainese et al., 2009). Drapes should be removed, heated humidifiers and hot air blowers should be turned off, and water mattresses should be turned to cooling temperatures. Room temperature normal saline solution should be given intravenously to maintain normal central venous pressure. The stomach can be irrigated with iced saline solution through an orogastric tube. Open body cavities can also be lavaged with iced saline solution, and ice packs can be placed in the groin and axillae where large vessels come close to the skin surface.
Hyperkalemia results when cell membranes are disrupted and when acidosis is severe. This is recognized on the electrocardiogram as increased T-wave amplitude in the early stages and later by widening QRS complexes, intraventricular conduction delays and blocks, and finally no organized rhythm at all. Intravenous calcium is appropriate emergency treatment of the hyperkalemia associated with MH (Gronert et al., 1986). Glucose and insulin (10 units of regular insulin in 50 mL of 50% glucose titrated to effect) can be administered to lower serum potassium temporarily. β-agonists can also be useful to move potassium intracellularly.
Large losses of intravascular volume should be anticipated; evaporative loss of fluid may be great, and edema formation may occur in muscles and in other tissues during fulminant MH. Intravenous fluids should be given to maintain normal cardiac filling pressures, as evidenced by adequate perfusion pressure, urine output, and capillary refill. Alkaline osmotic diuresis is induced by mannitol in the current formulation of dantrolene (150 mg of mannitol/mg of dantrolene in Dantrium). This protects renal tubule function in the presence of myoglobinuria and promotes acute intravascular volume loss. The management of the acute MH episode is summarized in Box 37-2.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


