FIGURE 14.1 Named branches of the tracheobronchial tree, viewed from the front. (From Nunn JF. Applied Respiratory Physiology. 3rd ed. London: Butterworths; 1987, with permission.)
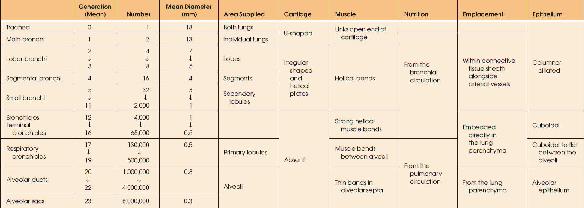
The primary lobular terminal respiratory unit is the likely equivalent of the alveolus when considered from the standpoint of function. The primary lobule is defined as the zone supplied by a first-order respiratory lobule. There are approximately 130,000 primary lobules with a diameter of about 3.5 mm containing approximately 2,000 alveoli each.
Alveolar ducts (generations 20 to 22) arise from terminal respiratory bronchioles and differ from terminal respiratory bronchioles by having no walls other than the mouths of mural alveoli (approximately 20 in number); approximately half of alveoli arise from ducts. The last generation of air passages differs from the alveolar ducts solely in the fact that they are blind pouches. Approximately 17 alveoli arise from these alveolar sacs and account for half of the total number of alveoli. Because of this increase in cross-sectional area resistance decreases and airflow slows down thus facilitating gas exchange over a large surface area.
| TABLE 14.1 Functional Anatomy of the Respiratory Tree |
 |
Alveoli
The total number of alveoli is approximately 300 million, but ranges from 200 to 600 million, corresponding to the height of the subject. The size of the alveoli is proportional to the lung volume; the alveoli are larger in the upper part of the lung due to higher negative pleural pressure, except at maximal inflation when the vertical size gradient disappears. The reduction in the size of alveoli and the corresponding reduction in the caliber of smaller airways in the dependent parts of the lung comprise the most important implications in gas exchange. During normal tidal breathing, most ventilator volume goes to lung bases. At functional residual capacity (FRC), the mean diameter is 0.2 mm (4).
Alveolar walls, which separate adjacent alveoli, consist of two layers of alveolar epithelium on a separate basement membrane enclosing the interstitial space. These layers contain pulmonary capillaries, elastin and collagen, nerve endings, and occasional neutrophils and macrophages. On one side of the interstitium, the capillary endothelium and alveolar epithelium are closely opposed, and the total thickness from gas to blood is usually less than 0.4 μm. This is the active side of the capillary, and gas exchange is more efficient at this site. The opposite side of the capillary is usually more than 1 to 2 μm thick and contains collagen and elastin fibers in an expanded tissue space. Herein is situated the connective tissue framework, which maintains pulmonary geometry. Alveolar septa are generally flat due to the tension generated by elastic fibers and surface tension at the air–fluid interface. The surface tension of the alveolar lining fluid is modified in the presence of surfactant, which decreases surface tension at the air fluid interface. Both elastin fibers and the decrease in surface tension keep the alveoli open during the volume change of the respiratory cycles so that gas exchange can continue. Septa are perforated by fenestrations known as pores of Kohn, which provide collateral ventilation; this can be demonstrated between the air spaces supplied by large bronchi (2).
Alveolar Cellular Morphology
The alveoli are divided by septa lined by flattened, continuous epithelial cells covering the thin interstitium (5). This epithelium, in humans, consists primarily of two distinct cells—type I and type II—with occasional neuroendocrine cells. In addition, although not frequently a part of the alveolar wall, the alveolar macrophage is, in fact, normally present on the alveolar epithelial surface.
Type I Epithelium
The type I alveolar cell (squamous lining cell), although comprising only 8% of parenchymal lung cells and inconspicuous by light microscopy, covers approximately 95% of the alveolar surface area, and has a total volume twice that of the histologically more prominent type II cell. Its nucleus is small and flattened, covered by a thin rim of cytoplasm containing few organelles. The remainder of the cytoplasm is aligned in broad sheets measuring 0.3 to 0.4 μm in thickness and extending in all directions for 50 μm or more over the alveolar surface. Sheets of adjacent type I cells interdigitate, and individual plates may reach into neighboring alveoli by winding the septal tip or by extending through the alveolar pores. Localized gap junctions have been identified between adjacent type I cells and between type I and type II alveolar cells frequently in association with an occluding junction (6).
The cytoplasm of type I epithelium contains few organelles but numerous pinocytotic vesicles, which are thought to transport fluid or proteins across the air–blood barrier. Type I cells have shown the ability to take up intra-alveolar particulate material and, while this particle clearance may be small in comparison with alveolar macrophages and the mucociliary apparatus, movement of materials across type I epithelium may allow particles to be deposited in regional lymph nodes.
Type II Epithelium
The type II epithelial cell (granular pneumocyte) is cuboidal in shape and protrudes into the alveolar lumen, making it easily identified on light microscopy. These cells may occur in groups of two or three and are often located near corners where adjacent alveoli meet. The cytoplasm of type II epithelium is rich in organelles, including endoplasmic reticulum with ribosomes, Golgi complexes, mitochondria, and membrane-bound osmiophilic granules. There is evidence from ultrastructural, biochemical tissue culture, and immunologic studies that type II cells and their osmophilic granules supply alveolar surfactant. These granules appear to function in a storage capacity, although some aspects of surfactant synthesis may also occur. Release of granule contents into the alveolar lumen occurs by exocytosis.
A second major function of type II epithelium is repopulation of normal and damaged alveolar epithelium. The type I cell is thought to be incapable of replication. On the other hand, the type II population is mitotically active and repopulates the alveolar surface. In addition, cytoplasmic simplicity and the large surface area of type I cells make them susceptible to damage from a variety of stimuli. In such circumstances, type II cells proliferate and temporarily repopulate alveolar walls, providing epithelial integrity. In time, they transform into type I cells. This sequence has been demonstrated with pulmonary injury from a variety of agents including oxygen, nitrous oxide, and other chemicals. Microvilli cover the surface of type II cells, suggesting that these cells may function in resorption of fluid or other materials from the alveolar air space.
Alveolar Macrophage
Pulmonary macrophages can be divided into three groups based on anatomic locations: (a) airway macrophage situated within the lumen or beneath the epithelial lining of conducting airways; (b) interstitial macrophage found isolated or in relation to lymphoid tissue in the interstitial connective tissue space; and (c) alveolar macrophage located on the alveolar surface. The alveolar macrophage has been the most extensively studied due to its accessibility by bronchoalveolar lavage (7).
The alveolar macrophage ranges from 15 to 50 μm in diameter and is round in shape with a foamy granular cytoplasm; nuclei are eccentric and may be multiple within the cell. Ultrastructurally, macrophages show prominent cytoplasmic projections that appear as microvillus-like structures. The cytoplasm contains a well-developed Golgi apparatus, scattered mitochondria, endoplasmic reticulum, ribosomes, microtubules and microfilaments, and membrane-bound granules of varying appearance. These granules contain primary and secondary lysosomes.
Pulmonary alveolar macrophages differ from other macrophages by having aerobic energy production, increased mitochondria and mitochondrial enzymes, and more numerous and larger lysosomes. Alveolar macrophages are ultimately derived from bone marrow precursors, presumably by way of the peripheral blood monocyte. In addition, there is evidence for a population of alveolar interstitial macrophages capable of division and replenishment or augmentation of the alveolar macrophage population in the absence of a functioning bone marrow or in times of increased stress. The average lifespan of a pulmonary macrophage in the air space is estimated at 80 days. Various inhaled toxins, including cigarette smoke, have a negative effect on macrophage viability and activity.
The functions of the alveolar macrophage are numerous; they are the primary phagocytes of the innate immune system, clearing the air spaces of infectious, toxic, or allergic particles that have evaded the proximal mechanical defenses. Alveolar macrophages also function as regulators of innate alveolar defenses against respiratory infection by synthesizing wide array of cytokines (including ILs-1, 6 and TNF-α), chemokines (IL-8), and arachidonic metabolites. Using these cell-to-cell signals, alveolar macrophages initiate inflammatory responses and recruit activated neutrophils in to the alveolar spaces. Recent evidence suggests that the alveolar macrophages have equally important role in resolving inflammation within the airspace. As the inflammatory response resolves, neutrophils undergo programmed cell death, or apoptosis. During apoptosis, neutrophil surface membranes remain intact, containing potentially injurious cytoplasmic contents. If apoptotic neutrophils are not efficiently cleared, leak of intracellular proteases into the alveolus from devitalized neutrophils produce further tissue injury and perpetuate inflammation. Efficient clearance of apoptotic neutrophils not only reduced macrophage secretions of proinflammatory cytokines but also stimulates production of anti-inflammatory cytokines, such as transforming growth factor-β and IL-10 (8).
Pulmonary Vasculature
Pulmonary Arterial and Venous Circulation
The pulmonary circulation carries the same flow as the systemic circulation, but arterial pressure and vascular resistance are normally one-sixth as great (2). The media of the pulmonary arteries are half as thick as in the systemic arteries of the corresponding size. In larger vessels, the media consist mainly of elastic tissue, but in smaller vessels, they are mainly muscular, with a transition being in vessels of 1 mm in diameter. Pulmonary arteries lie close to corresponding air passages in connective tissue sheaths.
The transition to arterioles occurs at an internal diameter of 100 μm. These vessels differ radically from the systemic circulation, as they are virtually devoid of muscular tissue. There is a thin medium of elastic tissue separated from blood by the endothelium. There is little structural difference between the pulmonary arterioles and venules.
Pulmonary capillaries arise from larger vessels—the pulmonary arterioles—and form a dense network over the walls of the alveoli; the spaces between them are similar in size to the capillaries themselves. About 75% of the capillary bed is filled in the resting state, but the percentage is higher in the dependent parts of the lung. This gravity-dependent effect is the basis of the vertical gradient of ventilation/perfusion ratios. Inflation of alveoli reduces the cross-sectional area of the capillary bed and increases the resistance to blood flow. Pulmonary capillary blood is collected into venules, which are structurally similar to arterioles. Unlike pulmonary arteries, pulmonary veins run close to the septa, which separate segments of the lung.
Bronchial Circulation
At the level of terminal bronchioles, air passages and accompanying blood vessels receive nutrition from bronchial vessels, which arise from systemic circulation. Part of this bronchial circulation returns to the systemic venous beds but mingles with pulmonary venous drainage, contributing to shunt. It has been established that when pulmonary arterial pressure in animals is raised as by massive pulmonary emboli, pulmonary arterial blood is able to reach pulmonary veins without traversing the capillary bed. This physiologic arteriovenous communication may offer an explanation for abnormalities of gas exchange during anesthesia.
Pulmonary Lymphatics
There are no lymphatics visible in the interalveolar septa, but small lymph vessels commence at the junction between the alveolar and extra-alveolar spaces. A well-developed lymphatic system courses around the bronchi and pulmonary vessels, capable of containing up to 500 mL of lymph, and draining toward the hilum (9). Down to airway generation 11, lymphatics lie in a potential space around air passages and vessels, separating them from lung parenchyma. This space becomes distended with lymph and pulmonary edema and accounts for the characteristic “butterfly shadow” (also termed “bat-wing appearance”) seen on a chest radiograph. In the hilum, lymphatic drainage passes through groups of tracheobronchial lymph nodes, where tributaries from superficial subpleural lymphatics contribute. Most of the lymph from the left lung enters the thoracic duct. Lymph from the right lung drains into the right lymphatic duct. Pulmonary lymphatics often cross the midline.
RESPIRATORY PHYSIOLOGY AND MECHANICAL VENTILATION
Positive pressure ventilation (PPV) as a life-sustaining measure first proved its merit during the polio epidemics of the 1950s. Since that time, the use of mechanical ventilatory (MV) support has been synonymous with the growth of critical care medicine. Early ventilation used neuromuscular blocking agents to provide control of patient respiratory efforts. Today, patient–ventilator interaction is critical, and there is a growing awareness of complications associated with neuromuscular blockade. Finally, there is increasing recognition that ventilators can induce various forms of lung injury, which has led to reappraisal of the goals of ventilatory support (10). While it seems that each manufacturer has introduced differing modes of MV, the fundamental principles of ventilatory management of critically ill patients remain unchanged.
PPV can be life saving in patients with hypoxemia or respiratory acidosis refractory to simpler measures. In patients with severe cardiopulmonary distress with excessive work of breathing, MV substitutes or supplements the action of respiratory muscles (11). In the setting of respiratory distress, respiratory muscles may account for as much as 40% of total oxygen consumption; in this circumstance, MV allows diversion of oxygen to other tissue beds that may be vulnerable. In addition, reversal of respiratory muscle fatigue, which may contribute to respiratory failure, depends on respiratory muscle rest. PPV can reverse or prevent atelectasis by allowing inspiration at a more favorable region of the pressure–volume curve describing pulmonary function. With improved gas exchange and relief from excessive respiratory muscle work, an opportunity is provided for the lungs and airways to heal. MV is not therapeutic in and of itself, and PPV may aggravate or initiate alveolar damage. These dangers of ventilator-induced lung injury have led to a reappraisal of the objectives of MV. Rather than seeking normal arterial blood gas values, it is often better to accept a degree of respiratory acidosis and possibly relative hypoxemia to avoid large tidal volumes and high inflation pressures.
MV may have hemodynamic effects as well. When applied to a passively breathing individual, PPV frequently lowers cardiac output (CO), primarily as a result of decreased venous return, especially when gas trapping occurs during passive inflation (12). In other circumstances, this form of ventilation may increase CO in the setting of impaired myocardial contractility because left ventricular afterload decreases with an increase in intrathoracic pressure. Alveolar distension compresses alveolar vessels, and the resulting increase in pulmonary vascular resistance and right ventricular afterload produces a leftward shift in the interventricular septum. Left ventricular compliance is decreased both by the bulging interventricular septum and increased juxtacardiac pressure from the distended lungs. There seems little doubt that adding MV or removing this support from critically ill patients can be a significant imposed stress.
MV strategies are clearly affected by underlying pulmonary disease. For example, in patients with acute respiratory failure, chronic obstructive pulmonary disease (COPD), asthma, or other conditions associated with a high residual volume, gas trapping develops in alveoli because patients have inadequate expiratory time available for exhalation before the next breath begins. Patients experiencing this “breath stacking” have a residual, peripheral positive end-expiratory pressure (PEEP). Also termed auto-PEEP, this retained peripheral gas makes triggering the ventilator more difficult, since the patient needs to generate a negative pressure equal in magnitude to the level of auto-PEEP in addition to the trigger threshold of the machine. This is one factor that may contribute to the patient’s inability to trigger the ventilator despite the obvious respiratory effort. Auto-PEEP may be undetected because it is not registered routinely on the pressure manometer of the ventilator, although newer machines have software to detect auto-PEEP. In older machines, occluding the expiratory port of the circuit at the end of expiration in a relaxed patient causes pressure in the lungs and the ventilator circuit to equilibrate, and the level of auto-PEEP is displayed on the manometer (13).
LUNG MECHANICS
Respiratory Muscles
Air flows to and from the alveoli, driven by differences in pressure between the airway opening and the alveolus. During spontaneous breathing, mouth (atmospheric) pressure remains constant, while alveolar pressure fluctuates under the influence of changing pleural pressure and tissue recoil forces (14–16). The diaphragm powers inspiration both by displacing the abdominal contents caudally and by raising the lower ribs, expanding them outward by a bucket handle effect (17,18). This latter action is aided by the external intercostal muscles. Normal exhalation is passive. When faced with a large ventilatory requirement or with impeded gas flow due to airway obstruction or parenchymal restriction, the accessory muscles of respiration are recruited to aid inhalation. Forceful exhalation is assisted by the internal intercostal muscles. The phrenic nerves (C3–C5) innervate the diaphragm, while the spinal nerves (T2–L4) innervate the intercostal and abdominal muscles.
The primary disorders of respiratory muscle function are usefully considered as problems of the diaphragm or problems of the accessory respiratory muscles (18,19). When upright, patients with isolated paralysis of both hemidiaphragms can often sustain adequate ventilation by the coordinated use of the intercostal and abdominal muscles. First, the diaphragm is forced upward as the muscles contract to raise the abdominal pressure. The diaphragm then descends, aided by gravity, as muscle relaxation allows abdominal pressure to fall. This mechanism cannot work effectively in the supine position, a circumstance that explains why orthopnea is a prominent symptom of this disorder. Patients with spinal cord injury (quadriplegia) have the converse anatomic problem: The intact diaphragm provides adequate ventilation to meet the normal requirement, but paralysis of the expiratory musculature severely limits ventilatory reserve and coughing efficiency.
Pressure–Volume Relationships
The lung and its thoracic shell occupy identical volumes, except when air or fluid separates them (20–22). At any specified volume, the pressure acting to distend the lung is alveolar pressure minus pleural pressure, while the pressure across the chest wall is pleural pressure minus atmospheric pressure. The volume of the lung is determined uniquely by lung compliance (distensibility) and the pressure difference acting to distend it (transpulmonary pressure). Thus, static lung volume is the same whether the alveolar pressure is 0 and pleural pressure is –5, or if alveolar pressure is 25 and pleural pressure is 20. A similar relationship between the distending pressure, compliance, and volume also applies to the chest wall. When the chest wall muscles are relaxed at FRC, the tendency of the chest wall to spring outward balances the tendency of the lung to recoil to a smaller volume; movement away from this equilibrium point requires muscular effort (Fig. 14.2). Should either the lung or the chest wall become less compliant (as in interstitial fibrosis or obesity), the pressure–volume curve shifts rightward and flattens, causing FRC to decrease (20). Conversely, an increased lung compliance (as in emphysema) allows a higher resting volume.

FIGURE 14.2 Static volume–pressure curves of the lung (PL), chest wall (PW), and total respiratory system (Prs) during relaxation in the sitting posture. The static forces of the lung and the chest wall are pictured by the arrows in the side drawings. The dimensions of the arrows are not to scale; the volume corresponding to each drawing is indicated by the horizontal broken lines. (From Vassilakopoulos T, Zakynthinios S, Roussos C. Muscle function: basic concepts. In: Marini JJ, Slutsky AS, eds. Physiological Basics of Ventilatory Support. New York: Marcel Dekker; 1998:114, with permission.)
Pleural Pressure
The fraction of change in alveolar pressure sensed in the pleural space depends on the relative compliances of the lung (CL) and chest wall (CW). For a given change in alveolar pressure (ΔPa), the amount transmitted to the pleural space (ΔPpl) will be:

An inherently stiff chest wall would allow no volume change of the lung and complete transmission of a given increment in alveolar pressure to the pleural space. Conversely, an infinitely stiff lung would transmit none of it. Under normal circumstances, the lung and chest wall are almost equally compliant throughout the tidal range, so that approximately half of any change in alveolar pressure (as when PEEP is applied) is recorded in the pleural space. In clinical practice, average pleural pressure is estimated for clinical purposes as esophageal pressure (23).
Although clinicians speak fondly of pleural pressure as if it were a unique number, pleural pressure varies considerably throughout the chest because of hydrostatic gradients (which at FRC averages 0.37 cm H2O/cm of vertical height). That translates in to higher pleural pressure (less negative) at lung bases due to weight of the lungs. At FRC, the average pleural pressure at midlung level is negative because the lungs are held open at greater than their relaxed volume. Pleural pressure surrounds the heart, the great vessels, and large airways, therefore affecting the vascular pressures measured at intrathoracic sites.
Effects of Changes in Lung Volume
Airway Resistance
Lung volume exerts a strong influence on airway resistance because resistance is inversely proportional to the fourth power of the radius of a conduit such as a bronchus. Pleural pressure surrounds the largest airways, while airways deeper within the lung are tethered open by the wall tension forces of the alveoli. Hence, as lung volume increases, the diameter of all airways increases, and resistance falls. Conversely, if a normal lung is held at a low resting lung volume, as in obesity, airway resistance will be high. In most restrictive diseases of lung tissue (e.g., interstitial fibrosis), the effects of heightened recoil on the airway diameter and driving force are usually more than sufficient to offset the effect of reduced volume, and flow rates are high relative to volume.
Pulmonary Vascular Resistance
Raising the lung volume has a different effect on the resistance of pulmonary vessels. Although the extra-2. 14.3).

FIGURE 14.3 Schematic representation of the effects of changes in vital capacity on total pulmonary vascular resistance and the contributions to the total afforded by alveolar and extra-alveolar vessels. During inflation from residual volume (RV) to total lung capacity (TLC), resistance to blood flow through alveolar vessels increases, whereas resistance through extra-alveolar vessels decreases. Thus, changes in total pulmonary vascular resistance form a U-shaped curve during lung inflation. FRC, functional residual capacity. (From Murray JF. Circulation. In: The Normal Lung: The Basis for Diagnosis and Treatment of Pulmonary Disease. Philadelphia: WB Saunders; 1976:131, with permission.)
Muscular Force
The lung volume has an important effect on the maximal inspiratory and expiratory muscular forces that can be generated. The magnitude of these forces can be quantified by measuring the pressure recorded against the occluded airway. At total lung capacity (TLC), the lung and chest wall exert their highest recoil pressures. More importantly, the muscles of expiration are stretched maximally and are able to generate their highest contractile forces. If the occluded airway port is suddenly released, as during coughing, intraluminal airway pressure falls. The flexible posterior walls of the central airways invaginate, and the lumen narrows markedly to a slit. As gas accelerates to a high velocity through this narrow region, it shears mucus from the airway walls and delivers it to the oropharynx. To be maximally effective, a coughing effort must be forceful and start from a high lung volume. Gas flows should not be obstructed in small airways, and the glottis must be sealed to allow pressure within the airway to build. In critically ill patients, all of these conditions may be violated simultaneously. For intubated patients, a vital capacity greater than 20 mL/kg and a maximal expiratory pressure of 60 mmHg against an occluded airway at TLC are good predictors of an effective cough post extubation. The greatest negative end-inspiratory pressure can be generated at a residual volume where the muscle fibers of the diaphragm are stretched maximally to a position of favorable mechanical advantage. This encourages some patients to unintentionally misuse incentive spirometers that place emphasis on achieving a high flow rate rather than a high inhaled volume; they often exhale below FRC in order to take advantage of higher inspiratory muscle efficiency (and the relatively minor tendency of the chest wall to spring outward) at lower volumes. Conversely, hyperinflation causes the diaphragm to work less effectively, adding to the sense of dyspnea experienced by patients with COPD (Fig. 14.3).
Position and Lung Volume
Position has an important influence on lung volume. In assuming a recumbent supine position, FRC falls approximately 25% to 30% (approximately 1,000 mL) in the adult, with most of the decrease occurring before the Fowler (30-degree) position (24). This reduction in lung volume occurs because the abdominal contents push the diaphragm upward. In either lateral recumbent position, the lung volume at FRC is only about 15% to 20% less than the upright sitting value because the nondependent (uppermost) lung maintains its sitting lung volume, or actually distends, partially offsetting the loss of volume from the lower lung. These observations have relevance for the nursing care of postoperative and critically ill patients.
Normal Pattern of Breathing
To provide fresh gas at 5 to 7 L/min to the lungs, the thoracic pump moves a stroke volume of 5 to 7 mL/kg at a frequency of 10 to 16 per minute. Once every 8 to 10 minutes, a sigh of two to four times the normal tidal volume occurs, which apparently serves to reverse the natural tendency for the individual alveoli to collapse when ventilated at a normal but monotonous volume. Breath-to-breath FRC changes continuously, at about a constant average value (25).
Dead Space
The bronchial, nasal, and pharyngeal passages do not participate in gas exchange. This anatomic dead space varies with airway caliber and lung volume, averaging roughly 2.2 mL/kg of lean body weight at FRC. Because approximately 50% of this dead space resides in the upper airways, orotracheal intubation and tracheostomy decrease anatomic dead space significantly (26). On the other hand, face masks and ventilator tubing unflushed by fresh gas can become an extension of the anatomic dead space, increasing the work of breathing. In addition to anatomic dead space, some volume of fresh gas (the alveolar dead space) reaches alveoli but does not participate in gas exchange because of inadequate perfusion. A portion of the increased ventilation requirement observed after a large pulmonary embolus results from this mechanism. Taken together, anatomic and alveolar dead space constitute the physiologic dead space—the volume of gas moved during each tidal breath that does not participate in gas exchange. The fraction of each tidal breath wasted in this fashion, the dead space volume-to-tidal volume (VD/VT) ratio, can be accurately approximated by the formula:
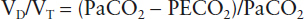
where PaCO2 and PECO2 are the partial pressures of CO2 in arterial blood and mixed expired gas, respectively. At a normal tidal volume, VD/VT increases with age; expressed as a percentage:

At very low tidal volumes, VD/VT rises to a high value because anatomic dead space does not decrease proportionately; nonetheless, even at tidal volumes theoretically below the anatomic dead space value, some alveolar gas exchange does occur. During exercise, the VD/VT may fall to 20% or less, owing both to large tidal breaths and better perfusion throughout the lung.
Flow Limitation
The rate of airflow depends on the pressure difference driving the flow and the resistance:
Flow = driving pressure/resistance
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


