Introduction to Pain Mechanisms: Implications for Neural Blockade
Philip J. Siddall
Michael J. Cousins
Pain is a complex phenomenon that has additional dimensions that set it apart from other sensations such as vision or hearing. The experience of pain, by nature, has an emotive, aversive component and arises from a multifaceted interaction of biological, psychological, and environmental contributors. In recent years, there has been a huge growth in our understanding of the details in each of these components and how they come together to contribute to the experience of pain. The aim of this chapter is not to provide a comprehensive review of the details of the different mechanisms and contributors to pain. More detailed information on the specific physiologic and pharmacologic processes of nociception, as well as the contribution of psychological factors, can be found in other chapters of this text (Chapters 32,33,34,35,36). Rather, this chapter will seek to provide a broad overview of our current concepts of pain and some of the implications that these concepts have for pain management and neural blockade.
What is Pain?
Before looking at some of these concepts, it may be helpful to examine what pain actually is. As mentioned, pain is a sensation that has an aversive quality. The International Association for the Study of Pain (IASP) has defined pain in this way: “Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage” (1). Inherent within this definition is the concept that pain is not only a sensory experience, but a complex interaction that also involves emotional and behavioral factors (Chapter 35).
Nervous System Plasticity
One of the fundamental shifts in our view of pain in the preceding century was the recognition of the adaptations that occur within the nervous system in response to pathophysiologic processes. At the beginning of the 20th century, many regarded pain as a sensation arising from the detection of a noxious stimulus by specific receptors, with transmission of signals along “hard-wired” nerve pathways specifically devoted to this process. However, by the end of the century, it was clear that this view was no longer tenable. Ample evidence now supports that the nervous system responds rapidly and often dramatically to changes in the environment and is in a continual state of flux that is dependent on the level of inputs arising from the periphery (2,3,4,5). For example, trauma to any part of the body, and nerve damage in particular, can lead to changes within other regions of the nervous system that influence subsequent responses to sensory input (6). Not only will an increase in inputs lead to a sensitization of central processing, but loss of inputs also appears to result in changes that are associated with the presence of persistent pain (7).
The gate theory, put forward by Melzack and Wall in 1965 (8), also attempted to deal with the problems associated with a hard-wired view of pain. Melzack and Wall proposed that an interaction of inputs occurs within the dorsal horn that can lead to an increase or decrease in the volume of signals transmitted through the dorsal horn to the brain, and that activity in larger primary afferents could inhibit messages traveling from small-diameter fibers. Although some of the details of the gate theory have not been substantiated, the gate theory did much to popularize the concept of modulation of sensory inputs at a spinal level. It also provided a theoretical framework for management approaches, such as stimulation techniques, that appeared to inhibit or “close the gate” on nociceptive inputs (see also Chapter 51).
The extension of the gate theory to include the influence of descending pathways from the brain went even further and helped to provide a framework for the inclusion of emotional and cognitive states in the experience of pain (9). This model set the groundwork for a more comprehensive approach to pain that recognized the contribution of sensory/discriminative, affective/motivational, and cognitive/evaluative components to pain perception and behavior. This model emphasizes the contribution of a person’s mood, cognitions and their external environment, and the importance of accurate identification and appropriate management of these contributing factors in the assessment and management of pain. The role and contribution of these psychological aspects to pain is described further in Chapter 35.
Pain in the Clinic
From a clinical point of view, pain is sometimes divided into physiologic and clinical entities (10). This division recognizes that, although many of the processes involved in nociceptive transmission have a role in the perception of acute noxious stimuli, different processes come into play when the stimulus is prolonged or sustained. Physiologic pain describes the situation
in which a noxious stimulus activates peripheral nociceptors, which then transmit sensory information through several relays until it reaches the brain and is recognized as a potentially harmful stimulus (Fig. 31-1, Table 31-1). In the clinical situation, however, this is rarely the situation. More commonly, the insult to the body that produces pain also results in inflammation and tissue injury. The pathophysiologic processes that occur following injury result in a stimulus–response pattern quite different from that seen following physiologic pain, and this phenomenon has therefore been termed pathophysiologic or clinical pain (Fig. 31-2, Table 31-2). It is the elucidation of these pathophysiological processes that has the most relevance for our understanding and management of pain in the clinical setting. Presentations of visceral pain differ clinically from somatic pain (Table 31-3).
in which a noxious stimulus activates peripheral nociceptors, which then transmit sensory information through several relays until it reaches the brain and is recognized as a potentially harmful stimulus (Fig. 31-1, Table 31-1). In the clinical situation, however, this is rarely the situation. More commonly, the insult to the body that produces pain also results in inflammation and tissue injury. The pathophysiologic processes that occur following injury result in a stimulus–response pattern quite different from that seen following physiologic pain, and this phenomenon has therefore been termed pathophysiologic or clinical pain (Fig. 31-2, Table 31-2). It is the elucidation of these pathophysiological processes that has the most relevance for our understanding and management of pain in the clinical setting. Presentations of visceral pain differ clinically from somatic pain (Table 31-3).
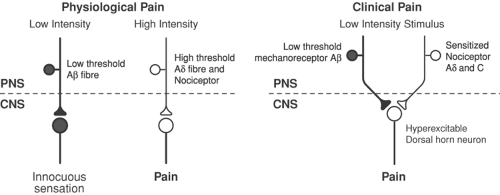 Figure 31-1. Right: In the clinical situation, central and peripheral changes lead to abnormal excitability in the nervous system. This means that low-intensity stimuli can produce pain. Left: Under “physiologic” conditions, low intensity, non-noxious stimuli activate low-threshold receptors to generate innocuous sensations, and high-intensity, noxious stimuli activate high-threshold nociceptors, which may lead to the sensation of pain. PNS, peripheral nervous system; CNS, central nervous system. From Woolf CJ, Chong MS. Pre-emptive analgesia-treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg 1993;77:362, with permission. |
Role of Genetic Factors in Pain
One of the questions raised in the clinical situation is why, in apparently identical situations, some people develop pain and others do not. For example, prevalence studies indicate that around 15% to 50% of people (depending on the condition) will develop neuropathic pain following nervous system trauma (11,12,13,14). This may be linked to physical factors, such as the nature of the injury, or to psychological factors that influence the perception and expression of pain. However, recent findings have also indicated the importance of our innate, genetic makeup in the way that we experience pain. Animal studies indicate that modifications to genetic information, such as occurs in transgenic and knockout mice, result in differences in the prevalence of nociceptive and neuropathic pain and differences in sensitivity to interventions and analgesics (15). Some researchers have suggested a genetic susceptibility to nociceptive and neuropathic pain with excessive and prolonged neuroplastic responses to neuropathic and nociceptive stimuli (16,17,18).
Table 31-1 Features of physiological pain | |
|---|---|
|
Some pain disorders have also been shown to have a genetic link. These include hereditary sensory neuropathy type 1 (19), painful congenital myotonia (20), and mutations of CACNL1A4 in people with familial hemiplegic migraine (21) or the neurotrophin receptor tropomyosin receptor kinase A (trkA) in people with congenital insensitivity to pain (22).
μ-Opioid receptor gene polymorphisms (Oprm) (23) cause abnormalities of sensation or changes in response to medication (e.g., differing CYP2D6 phenotype) (24).
μ-Opioid receptor gene polymorphisms (Oprm) (23) cause abnormalities of sensation or changes in response to medication (e.g., differing CYP2D6 phenotype) (24).
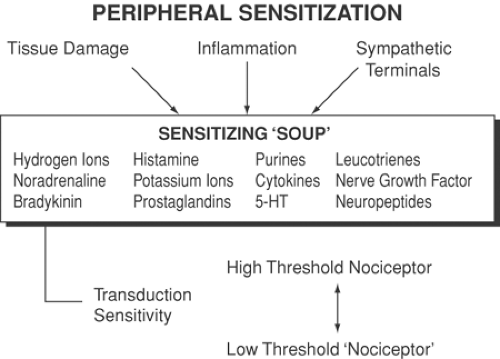 Figure 31-2. The sensitivity of high-threshold nociceptors can be modified in the periphery by a combination of chemicals that act as a “sensitizing soup.” These chemicals are produced by damaged tissue as part of the inflammatory reaction and by sympathetic terminals. 5-HT, 5-hydroxytryptamine. From Woolf CJ, Chong MS. Pre-emptive analgesia-treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg 1993;77:362, with permission. |
Table 31-2 Features of clinical pain | |
|---|---|
|
An important link between pain and genetic factors has been demonstrated in a study that found that pain responses are modified by alterations in the concentration of an enzyme cofactor, tetrahydrobiopterin (BH4) (25). This study also found that radicular pain in patients following diskectomy was significantly less in people who had a haplotype of the GTP cyclohydrolase gene, which results in reduced production of BH4. This study suggests a strong link between genetic makeup and the development of neuropathic pain. Thus, although the complexity of pain suggests that it is extremely unlikely that a single “pain gene” will be discovered, nevertheless genetic factors are clearly involved in the development of pain under certain conditions and are involved in response to treatment (see also Chapter 33).
Pain as a Disease Entity
Pain has often been regarded merely as a symptom that serves as a passive warning signal of an underlying disease process. Using this model, the goal of treatment has been to identify and address the pathology causing pain in the expectation that this would lead to the resolution of pain. However, it has been suggested that it is more appropriate to consider persistent pain as a disease entity (26,27). As this and the following three chapters demonstrate, accumulating evidence suggests that the transmission of nociceptive signals following injury has a rapid and sustained impact on the physiologic environment at a number of levels (4,6,28,29,30). In addition, pain has a profound impact on psychological function and social relationships (Chapter 35). These processes, associated with persistent nociceptive inputs, range from changes in receptor function to mood dysfunction, inappropriate cognitions, and social disruption. Therefore, it can be argued that simply regarding pain as a symptom is deficient and fails to recognize the secondary pathophysiologic processes that occur as a result of nociceptive inputs. It also minimizes the impact of pain on the psychological and social milieu of the person in pain. These changes that occur as a consequence of continuing nociceptive inputs and that are described herein argue for the consideration of persistent pain as a disease entity in its own right.
Table 31-3 Visceral pain compared with somatic pain | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pain Mechanisms
Peripheral Mechanisms
Primary Afferent Nociceptors
The primary afferent nociceptor is generally the initial structure involved in nociceptive processes. Most body structures contain nerve endings that are responsive to mechanical, thermal, and chemical stimuli (see Chapter 30). Nociceptors are primary afferent nerve fibers that are capable of encoding noxious stimuli. Depending on the response characteristics of the nociceptor, stimulation results in propagation of impulses along the primary afferent nerve fiber toward the spinal cord. The receptors associated with transmission of noxious information can be grouped into two main categories: Aδ-fiber mechanothermal and C-fiber polymodal nociceptors (see Chapter 32). Although Aβ-fibers can respond to noxious stimuli, they do not increase their response with increasing stimulus strength and therefore do not encode stimuli in the noxious range.
Receptors
A variety of receptors on primary afferent nociceptors transduce sensory stimuli into nerve activity. These include acid-sensing ion channels (ASIC) (31), P2X receptors (sensitive to adenosine triphosphate [ATP]) (32), and a number of transient receptor potential (TRP) channels such as the cold- and menthol-sensitive transient receptor potential menthol (TRPM8) channel and the heat- and capsaicin-sensitive
transient receptor potential vanilloid (TRPV1) channel (33). The TRPV1 receptor responds to heat and lowering of pH and may help mediate the increased pain and hypersensitivity that occurs with inflammation (34) (Fig. 31-3).
transient receptor potential vanilloid (TRPV1) channel (33). The TRPV1 receptor responds to heat and lowering of pH and may help mediate the increased pain and hypersensitivity that occurs with inflammation (34) (Fig. 31-3).
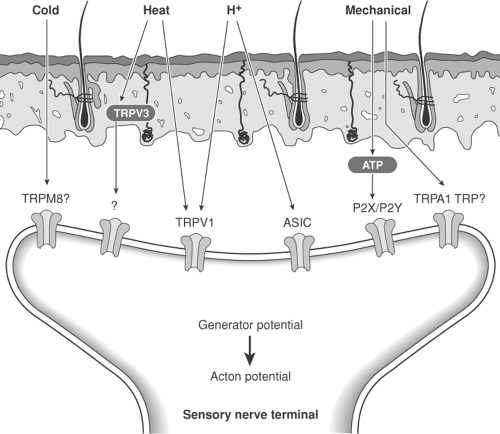 Figure 31-3. Peripheral transduction mechanisms. A major advance has resulted from molecular biology studies of nociceptive transduction mechanisms. Noxious mechanical stimuli release adenosine triphosphate, which may act on one or more purine receptors (P2X and P2Y). A second mechanism is still uncertain and may involve transient receptor potential (TRP)A1 or other TRP receptors. Noxious chemical stimuli such as acidity (H+) act via acid-sensing ion channels (ASICs) or TRPV1 receptors. Noxious heat acts via TRPV1 and TRPV2 (originally called VRL-1) possibly via TRPV3 receptors. Noxious cold acts via TRPM8 receptors (previously called CMR1). VRL, vanilloid receptor–like; CMR, cold menthol receptor. Modified from Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci 2005;6: 521–532, with permission. |
Visceral Afferents
Most afferent nerve fibers arising from visceral structures are free nerve endings (35). However, some organs, such as the liver, brain, or lung, do not appear to have sensory innervation and therefore do not respond to traumatic stimuli. The detection of stimuli by visceral structures also appears to be different, and although many structures may be poor in detecting mechanical stimulation such as cutting, they do respond vigorously to other stimuli such as distension. As well as different response properties, visceral afferent pathways are also distinctive from those involved in the transmission of sensory information from somatic structures. Visceral pain is characteristically diffuse and poorly localized (Table 31-3). Like somatic pain, central sensitization may occur in response to an initial insult and result in pain that appears to be out of proportion to the underlying pathology. An example of this is irritable bowel syndrome, which is believed to have a central component leading to a heightened response to relatively minor stimuli. Central sensitization may also be an important component of pain associated with clearly identified pathology, such as ureteric calculi or urinary tract infections, and lead to an exaggeration of response to the underlying pathology.
Referral of visceral pain is also a commonly recognized clinical phenomenon. The mechanism of referral is still a matter of debate but one suggested mechanism is convergence. This term refers to the convergence of terminals from visceral and somatic nociceptors onto the same neurons that ascend from the spinal cord to the brain. This makes it difficult for the brain to distinguish whether signals are arising from somatic or visceral structures (see below Fig. 31-21).
Peripheral Sensitization
Many forms of pain arise from direct activation of primary afferent neurons, especially C-fiber polymodal nociceptors (36). However, the process of nociceptor activation sets in train other processes that modify and enhance responses to further stimuli, a process collectively known as peripheral sensitization. For example, a relatively benign noxious stimulus such as a scratch to the skin initiates an inflammatory response in the periphery, which then changes the response properties to subsequent sensory stimuli (Fig. 31-4). After peripheral sensitization, low-intensity mechanical stimuli that would not normally cause pain are now perceived as painful. An increased responsiveness to thermal stimuli also occurs at the site of injury. This zone of primary hyperalgesia surrounding the site of injury is due to peripheral changes and is a feature commonly observed following surgery and other forms of trauma.
Part of the inflammatory response is the release of intracellular contents from damaged cells and inflammatory cells such as macrophages, lymphocytes, and mast cells. Nociceptive stimulation also causes a neurogenic inflammatory response, with the release of substance P, neurokinin A, and calcitonin gene-related peptide (CGRP) from the terminals of nociceptive afferent fibers in the periphery (37). Release of these peptides results in a changed excitability of sensory and sympathetic nerve fibers, vasodilatation, and extravasation of plasma proteins, as well as acting on inflammatory cells to release chemical mediators (38,39,40). These interactions induce release of a “soup” of inflammatory mediators such as potassium, serotonin, bradykinin, substance P, histamine, cytokines, nitric oxide (NO), and products from the cyclooxygenase and
lipoxygenase pathways of arachidonic acid metabolism (Fig. 31-5) (39,40,41).
lipoxygenase pathways of arachidonic acid metabolism (Fig. 31-5) (39,40,41).
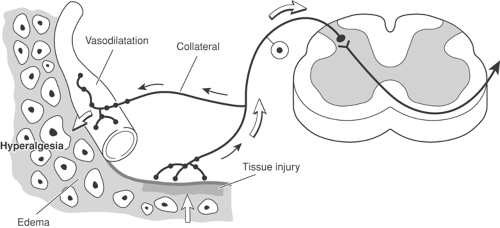 Figure 31-4. Effect of antidromic impulses in primary afferents. After tissue injury, antidromic impulses in collaterals of primary afferent fibers results in the release of chemical mediators from the peripheral terminals, which produce vasodilatation (flare), edema (wheal), and hyperalgesia (triple response of Lewis). Modified from Fields HL. Pain. New York, McGraw-Hill, 1987, with permission. |
As well as release of this soup of chemicals, inflammation also results in other changes that may contribute to peripheral sensitization. Neurotrophins act as pain modulators and have an important role in regulating neural function as well as structure (42,43). Nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) are two neurotrophins that have received considerable attention. Nerve growth factor acts on the tyrosine kinase A (trkA) receptor and is a peripheral pain mediator that is upregulated in inflammatory conditions. It has numerous actions, which include upregulation of neuropeptides such as CGRP and substance P, receptors such as TRPV1 and P2X3, and ion channels such as tetrodotoxin (TTX)-sensitive or -insensitive sodium channels (33). These actions may all contribute to sensitization of the primary afferent fibers. Modulation of peripheral sensitization may result from peripheral release of opioids from inflammatory cells (Fig. 31-6). Inflammation and peripheral sensitization may also lead to the recruitment of nociceptors that, under normal conditions, are silent. These “silent nociceptors” have been identified in a number of different tissues and species and once again bear testimony to the dynamic nature of sensory function. They are a class of unmyelinated primary afferent neurons that do not respond to excessive mechanical or thermal stimuli under normal circumstances (44). However, in the presence of inflammation and chemical sensitization, they become responsive, discharging vigorously even during ordinary movement and displaying alterations in receptive fields (45).
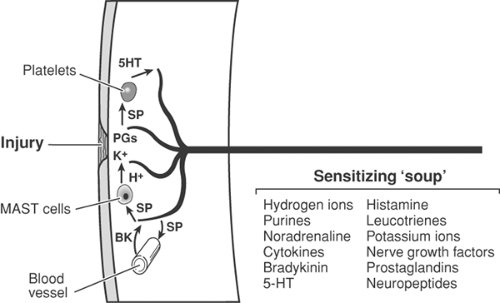 Figure 31-5. Peripheral sensitization following injury. Injury results in release of potassium (K+) from damaged cells, as well as prostaglandins. K+ strongly activates nociceptive free nerve endings (Aδ- and C-fibers). Prostaglandins sensitize nociceptors to activators such as K+. Antidromic stimuli in collaterals (see Fig. 31-4) activate release of substance P (SP) (and calcitonin-gene-related peptide [CGRP]; not shown), which plays a pivotal role at three sites: SP (and CGRP) increase capillary permeability, thus allowing the peptide bradykinin to cross capillary walls and strongly activate nociceptors, in turn releasing more SP; SP acts on platelets to release 5-hydroxy tryptamine (5HT), which sensitizes nociceptors, again releasing more SP; SP acts also on mast cells to release histamine, sensitizing nociceptors. The foregoing three SP processes set up vicious circles of increasing sensitization. PGs. prostaglandins. |
Peripheral Nerve Injury
Primary afferent nerves are not simply inert conductors of sensory information. Studies have demonstrated that section of, or damage to, a peripheral nerve results in a number of biochemical, physiologic, and morphologic changes that act as a focus of pain in themselves (Fig. 31-7) (30). The damaged end of the nerve fiber sprouts and may produce a spontaneously firing neuroma (Fig. 31-8). It may also demonstrate changed properties in response to various stimuli. These properties include sensitivity to mechanical stimuli, spontaneous firing, and sensitivity to norepinephrine (noradrenaline) (30). Similar changes
occur within the cell body of the primary afferent nociceptor, the dorsal root ganglion (DRG) (46). Reduction in the blood supply to myelinated fibers ends in demyelination and the production of ectopic impulses.
occur within the cell body of the primary afferent nociceptor, the dorsal root ganglion (DRG) (46). Reduction in the blood supply to myelinated fibers ends in demyelination and the production of ectopic impulses.
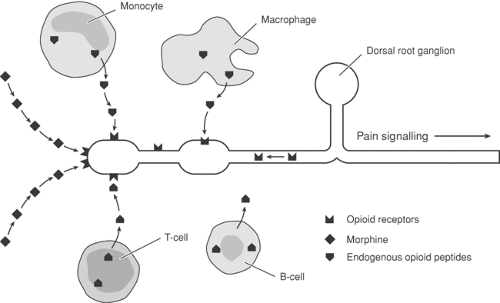 Figure 31-6. Peripheral inflammation results in production of opioid receptors by the dorsal root ganglion (DRG) and transport of opioid receptors toward the peripheral terminal. Peripheral opioid receptors are activated by exogenous application of morphine and endogenous opioid peptides released by monocytes (M), T-cells (T), B-cells (B), and macrophages (MP). Modified from Stein C. Morphine: A local “analgesic.” Pain: Clinical Updates 1995;3:1, with permission. |
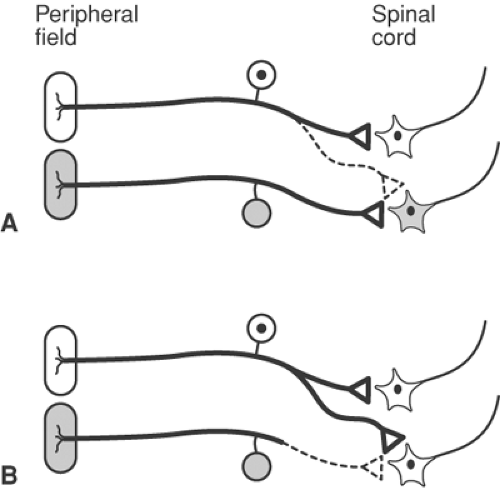 Figure 31-7. Neuroplasticity following damage to primary afferent fibers. A: Normal connectivity of primary afferent. Primary afferents innervate a defined peripheral region and activate a specific population of spinal cord neurons. In addition, the primary afferent has central connections that are normally ineffective (dashed line). In the normal situation, each spinal cord cell responds only to stimulation of its own peripheral field. B: When the central process of a primary afferent that innervates an adjacent peripheral field (stipple) is interrupted (dotted line), the formerly ineffective central connection of the intact primary afferent (heavy line) becomes effective. Both spinal cord cells now respond only to stimulation of the innervated peripheral field (not stippled). Modified from Fields HL. Pain. New York, McGraw-Hill, 1987, with permission. |
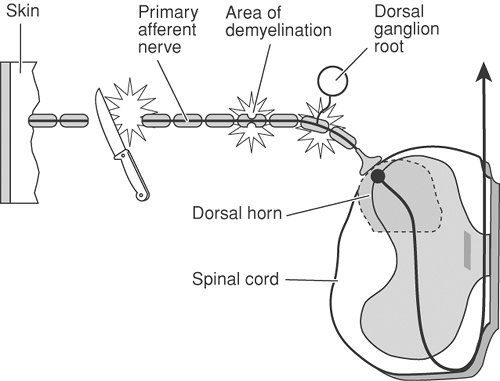 Figure 31-8. Sites of ectopic discharge in damaged primary afferent nociceptors. A transected nerve begins to regenerate, sending out sprouts that are mechanically sensitive, sensitive to α-adrenergic agonists, and spontaneously active. In addition, a secondary site of hyperactivity develops near the cell body in the dorsal root ganglion. Ectopic impulses may also arise from a short patch of demyelination on a primary afferent. From Fields HL. Pain. New York, McGraw-Hill, 1987, with permission. |
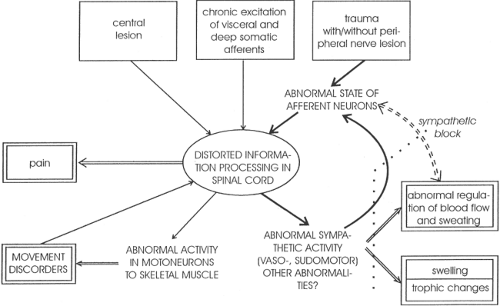 Figure 31-9. General hypothesis about the neural mechanisms of the generation of complex regional pain syndrome (CRPS) I and II following peripheral trauma, with and without nerve lesions, chronic stimulation of visceral afferents (e.g., myocardial infarction) and deep somatic afferents and, rarely, central trauma. The clinical observations are double-framed. Note the vicious circle (arrows in bold black). An important component of this circle is the excitatory influence of postganglionic sympathetic axons on primary afferent fibers in the periphery. From Jänig W. The puzzle of “reflex sympathetic dystrophy”: Mechanisms, hypotheses, open questions. In: Jänig W, Stanton-Hicks M, eds. Reflex Sympathetic Dystrophy: A Reappraisal. Seattle, IASP Press, 1996, with permission. |
A number of receptor changes may underlie this increased sensitivity and ectopic activity in primary afferent fibers. Peripheral nerve injury leads to the abnormal expression of receptors such as α2-adrenergic receptors, calcium channels (47), and novel TTX-sensitive or -insensitive sodium channels (48,49,50). Novel sodium channel expression may be a source of abnormal ectopic discharges from neuromata and the DRG (51,52) of both injured and uninjured (53) primary afferents. Nerve damage also results in an increased production of neurotrophins such as NGF and BDNF. This upregulation of NGF and BDNF may contribute to the development of pain through both peripheral and central effects (42,43).
Sympathetic Nervous System
The sympathetic nervous system also has an important role in the generation and maintenance of chronic pain states (54,55). Nerve damage and even minor trauma can lead to a disturbance in sympathetic activity (Fig. 31-9), which then leads to a sustained condition now termed a complex regional pain syndrome (CRPS), replacing the previously used terms reflex sympathetic dystrophy and causalgia (1,56). Complex regional pain syndromes are associated with features of sympathetic dysfunction, including vasomotor and sudomotor changes, abnormalities of hair and nail growth, osteoporosis, sensory symptoms of spontaneous burning pain, hyperalgesia and allodynia, and, often, disturbance of motor function (54,56,57,58).
Basic studies have demonstrated that several changes involving the sympathetic nervous system may be responsible for development of these features (59,60,61). Inflammation can result in the sensitization of primary nociceptive afferent fibers by prostanoids that are released from sympathetic fibers (62) (Fig. 31-10). Following nerve injury, sympathetic nerve stimulation or administration of norepinephrine can excite primary afferent fibers via an action at α-adrenoceptors (63). There is also innervation of the DRG by sympathetic terminals (59). This means that activity in sympathetic efferent fibers can lead to abnormal activity or responsiveness of the primary afferent fiber.
Complex regional pain syndromes may be sympathetically maintained or sympathetically independent. Pain problems that are sympathetically maintained may respond to sympathetic blockade by agents administered systemically, epidurally, regionally, or around the sympathetic ganglion (64) (see Chapter 39). Further information on the features, mechanisms, and treatment of CRPS can be found in Chapter 46.
Spinal Mechanisms
Termination Sites of Primary Afferents
The dorsal horn is the site of termination of primary afferents and there is a complex interaction among primary afferent fibers, local intrinsic spinal interneurons, and the endings
of descending fibers from the brain (65) (Chapter 32 and Figure 31-18). Primary afferent nociceptors terminate primarily in laminae I, II (substantia gelatinosa), and V (66), where they connect with several classes of second-order neurons in the dorsal horn of the spinal cord. Large fibers transmitting innocuous inputs primarily terminate in laminae III and IV. Some fibers ascend and descend several segments in the Lissauer tract before terminating on neurons that project to higher centers (see Chapter 32).
of descending fibers from the brain (65) (Chapter 32 and Figure 31-18). Primary afferent nociceptors terminate primarily in laminae I, II (substantia gelatinosa), and V (66), where they connect with several classes of second-order neurons in the dorsal horn of the spinal cord. Large fibers transmitting innocuous inputs primarily terminate in laminae III and IV. Some fibers ascend and descend several segments in the Lissauer tract before terminating on neurons that project to higher centers (see Chapter 32).
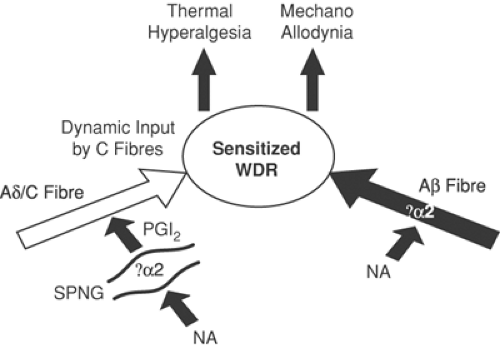 Figure 31-10. Inflammation and nerve injury leads to sensitization of wide dynamic range (WDR) neurons in the dorsal horn. This process involves sensitization of nociceptive primary afferents by release of prostanoids (PGI2) from sympathetic fibers and activation of Aβ-fiber α2-adrenoceptors by noradrenaline (NA). The effect of these interactions is a resultant thermal hyperalgesia and mechanical allodynia. |
Two main classes of second-order dorsal horn neurons are associated with sensory processing. The first class of neurons is termed nociceptive-specific or high-threshold; the second class is termed wide dynamic range or convergent. The two classes have different response properties to afferent input and are located in different regions of the dorsal horn. Nociceptive-specific neurons are located within the superficial laminae (I, II) of the dorsal horn and respond selectively to noxious stimuli (67). Wide dynamic range neurons generally are located in deeper laminae (V) and respond to both noxious and non-noxious input (68). Wide dynamic range neurons normally do not signal pain in response to a tactile stimulus at a non-noxious level. However, if they become sensitized and hyperresponsive, they may discharge at a high rate following a tactile stimulus (see Chapter 32). If the activity of the wide dynamic range neuron exceeds a threshold level following this stimulus, then the non-noxious tactile stimulus will be perceived as painful and give rise to the phenomenon of allodynia (69).
Neurotransmitters
Pharmacologic studies have helped to identify the many neurotransmitters and neuromodulators involved in pain processes in the dorsal horn (70) (see Chapter 33). The main neurotransmitter is the excitatory amino acid glutamate. Primary afferent nociceptors are also sometimes divided according to their neurotransmitter content. One group is peptidergic neurons that contain peptides such substance P, neurokinin A, and CGRP. Substance P and neurokinin A act on neurokinin 1 and neurokinin 2 receptors respectively. The other group of nonpeptidergic neurons express the P2X3 purinergic receptor. A number of other receptors are also involved in nociceptive transmission or modulation in the dorsal horn; they include opioid, α-adrenergic, cholinergic, γ-aminobutyric acid (GABAergic), glycinergic, serotonergic (5HT), and tyrosine kinase (trk) receptors (Figs. 31-11 and 31-12).
Excessive neuronal firing may induce prolonged glutamate release and activation of metabotropic glutamate (mgluR)and ionotropic N-methyl-D-aspartate (NMDA) receptors, with subsequent calcium influx into second-order neurons (71). The excitatory amino acids act at NMDA receptors, non-NMDA receptors (such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AMPA), kainate, and metabotropic glutamate receptors (70,72) (Figs. 31-11 and 31-12).
N-Methyl-D-Aspartate Receptor
A longstanding interest has existed in the role of NMDA receptor in the development of central sensitization, and this receptor appears to be a major contributor to this process. The NMDA receptor channel, in its resting state, is blocked by a magnesium “plug.” Priming of the NMDA receptor through the continued release of glutamate and coactivation of neurokinin receptors leads to removal of the magnesium plug and subsequent calcium influx into the cell (Figs. 31-12 and 31-13). Calcium influx, in concert with activation of other receptors on the cell membrane, precipitates activation or production of a number of intracellular messengers or signaling molecules, including phospholipases, polyphosphoinosites (IP3, DAG), ERK, NO, cyclic AMP response element-binding protein (CREB), and protein kinases (73,74,75,76) (Figs. 31-11,31-12,31-13,31-14,31-15). These intracellular messengers then act directly to change the excitability of the cell by feeding back and directly enhancing receptor activation or by feeding forward and inducing gene transcription within the cell nucleus (77). Gene transcription then results in receptor changes that lead to long-term changes in the responsiveness of the cell. All of these processes contribute to the development of central sensitization and some of the medium- and long-term alterations in cellular responsiveness that underlie chronic pain states (78,79) (Figs. 31-11 and 31-15).
The exact role of NO in nociceptive processing is still unclear, and it does not appear to be important in acute nociception (75,80,81). However, production of NO is implicated in the induction and maintenance of chronic pain states (75), and may be one of the factors responsible for the cell death demonstrated to occur under these conditions (Fig. 31-14). The production of arachidonic acid metabolites as part of the cascade that occurs following NMDA receptor activation also raises an interesting potential avenue for intervention that has been explored (82). Although the peripheral effects of nonsteroidal anti-inflammatory drugs (NSAIDs) have been emphasized in the past, it appears that there may be a role for the spinal administration of NSAIDs. Spinal NSAIDs either act directly on receptors, such as the strychnine-insensitive glycine site of the NMDA receptor complex, or influence the production of metabolites within the cell (Figs. 31-11 and 31-13).
Other Receptors
Although much research has focused on the NMDA receptor, as mentioned previously, a number of receptors are involved in nociceptive transmission, such as the metabotropic glutamate, neurokinin, tyrosine kinase (trk), AMPA, and kainate receptors (83,84). These receptors also contribute to the development of central sensitization either as a result of ion influx or activation of intracellular signaling cascades. Neurotrophins also contribute to central sensitization. Brain-derived neurotrophic factor production is upregulated in inflammatory conditions and is released from the central terminations of primary afferents in the dorsal horn, where it acts as a pain modulator. It acts on the tyrosine kinase B (trkB) receptor, which activates signaling cascades within dorsal horn neurons that contribute to the development of central sensitization (42) (Fig. 31-11A).
As well as directly increasing excitability, sensitization may also occur through a reduction in normal inhibitory processes. This may occur as a result of changes in the release of GABA or changes in receptor function. It has been suggested that prolonged stimulation, presumably through sustained and therefore excitotoxic release of glutamate, may result in the death of GABAergic inhibitory neurons and thereby lead to amplification of sensory inputs (85,86). In addition, alterations may occur in GABAergic function. For example, BDNF may act as a signaling molecule that allows communication between glia and neurons and contributes further to central sensitization by reversing the normal inhibitory function of GABA within the dorsal horn (87,88) (Fig. 31-11).
Glia
Glia (microglia and astrocytes) have traditionally been regarded as having a predominantly supportive role in neural signaling, and much of pain research has focused on the function and role of neurons in pain processing. However, abundant evidence now suggests the importance of glia in pain transmission and the development and maintenance of pathologic pain conditions (89,90). Glia are known to be activated by viruses and bacteria, but they also respond to substances released by
primary afferent nerve fibers such as substance P, glutamate, and neurotrophins. Activated glia in turn release a variety of neuroactive chemicals such as cytokines, glutamate, NO, and ATP. These substances can then act to enhance release of chemicals from primary afferent terminals or sensitize pain transmission neurons within the dorsal horn (see Chapter 32).
primary afferent nerve fibers such as substance P, glutamate, and neurotrophins. Activated glia in turn release a variety of neuroactive chemicals such as cytokines, glutamate, NO, and ATP. These substances can then act to enhance release of chemicals from primary afferent terminals or sensitize pain transmission neurons within the dorsal horn (see Chapter 32).
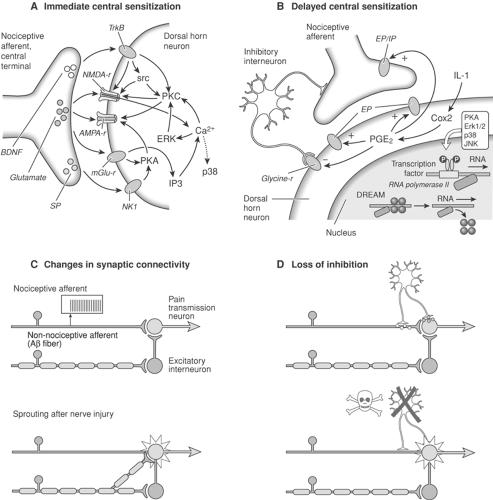 Figure 31-11. Non-nociceptor–mediated pain is generated by sensory inputs that would normally produce an innocuous sensation; this reflects a change in the functioning of central neurons. A: Activity-dependent central sensitization. An immediate and relatively short-lasting increase in the excitability and responsiveness of pain transmission dorsal horn neurons, which is due to phosphorylation of ion channels and receptors and follows nociceptor-driven transmitter release and activation of intracellular kinases. Eventually, the response to normally subthreshold inputs is increased. B: Transcription-dependent central sensitization. Enhanced gene expression due to the activation of transcription factors, as well as the removal of repressors like downstream regulatory element antagonist modulator (DREAM), results in long-lasting changes in the function of dorsal horn neurons. Cyclooxygenase inhibitor (COX2) induction leads to prostaglandin (PGE2) production, which acts pre- and postsynaptically to facilitate excitatory and reduce inhibitory transmission. C: After peripheral nerve injury, the central terminals of myelinated non-nociceptive Aβ-afferents sprout in the dorsal horn and form new connections with nociceptive neurons in laminae I and II. This rewiring of the circuitry of the spinal cord may contribute to persistent pain hypersensitivity. D: Disinhibition. Normal sensory inflow is actively controlled by inhibitory interneurons. Reduced synthesis of the inhibitory neurotransmitters γ-aminobutyric acid (GABA) and glycine or loss of these inhibitory interneurons after excessive release of the excitotoxic amino acid glutamate following peripheral nerve injury increases the excitability of pain transmission neurons such that they begin to respond to normally innocuous inputs. Modified from Nature Neuroscience 2002;5:1065, with permission. |
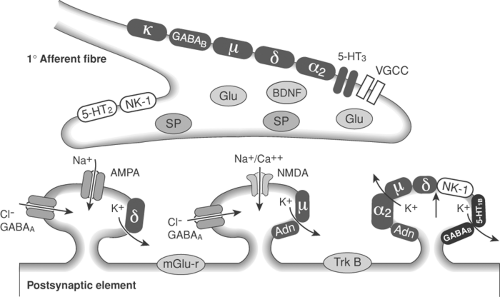 Figure 31-12. Possible arrangement of receptors on pre- and postsynaptic structures in the dorsal horn of the spinal cord. 5-HT, serotonin; α2, α2-adrenoceptor; Adn, adenosine; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; GABA, γ-aminobutyric acid; Glu, glutamate; NMDA, N-methyl-D-aspartate; NK-1, neurokinin 1; μ, δ, κ, opioid receptors; SP, substance P. BDNF, brain-derived neurotrophic factor; VGCC, voltage-gated calcium channel; mGlu-r, metabotrophic glutamate receptors; TrK B-tyrosine kinase B receptor. |
Central Sensitization
As described earlier, it is known that changes occurring in the periphery following trauma lead to an enhanced responsiveness of dorsal horn neurons to further stimuli and a zone of primary hyperalgesia at the site of injury. However, this enhanced responsiveness can only partly be explained by the changes in the periphery. Following injury, there is an increased responsiveness to normally innocuous mechanical stimuli (allodynia) in a zone of secondary hyperalgesia in uninjured tissue surrounding the site of injury. In contrast to the zone of primary hyperalgesia, no change occurs in the threshold to thermal stimuli. These behavioral changes are believed to be a result of the dorsal horn processes (described earlier) that contribute to the development of central sensitization (Fig. 31-16) (91). Thus, central sensitization refers to the increased responsiveness of neurons in the spinal cord that occurs following strong and sustained inputs from the periphery.
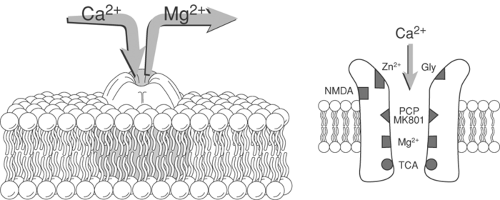 Figure 31-13. Representation of the N-methyl-D-aspartate (NMDA) receptor complex. Removal of the magnesium plug leads to calcium influx. The NMDA receptor complex contains a number of sites, activation of which results in modulation of receptor activity. These sites include: glycine (GLY), dizocilpine (MK-801), phencyclidine (PCP), magnesium (Mg2+), tricyclic antidepressant (TCA), and zinc (Zn2+). |
These changes are important in the consideration of clinical pain. A barrage of nociceptive inputs, such as occurs with surgery, results in changes to the response properties of dorsal
horn neurons (92). It has been demonstrated that a painful stimulus at a level sufficient to activate C-fibers not only activates dorsal horn neurons, but neuronal activity also progressively increases throughout the duration of the stimulus, a phenomenon termed wind-up (93). Therefore, with continued nociceptive inputs, a simple stimulus–response relationship does not exist, but rather a gradual increase in spinal cord neuronal activity (see Chapter 32). Wind-up is dependent on activation of the NMDA receptor (71,94) and therefore has the potential to be modified by agents acting at this site. Thus, wind-up may make these neurons more sensitive to other input and is a component of central sensitization (Figs. 31-16 and 31-17).
horn neurons (92). It has been demonstrated that a painful stimulus at a level sufficient to activate C-fibers not only activates dorsal horn neurons, but neuronal activity also progressively increases throughout the duration of the stimulus, a phenomenon termed wind-up (93). Therefore, with continued nociceptive inputs, a simple stimulus–response relationship does not exist, but rather a gradual increase in spinal cord neuronal activity (see Chapter 32). Wind-up is dependent on activation of the NMDA receptor (71,94) and therefore has the potential to be modified by agents acting at this site. Thus, wind-up may make these neurons more sensitive to other input and is a component of central sensitization (Figs. 31-16 and 31-17).
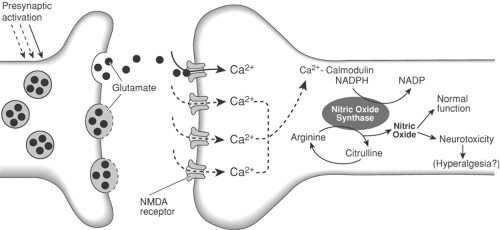 Figure 31-14. Diagram illustrating postsynaptic events following release of glutamate from central terminals of primary afferents in the spinal cord. Following priming of the N-methyl-D-aspartate (NMDA) receptor complex, subsequent glutamate release results in NMDA receptor activation with subsequent calcium influx. Intracellular calcium then acts on a calmodulin-sensitive site to activate the enzyme nitric oxide synthase (NOS). In the presence of a cofactor nicotinamide adenine dinucleotide phosphate (NADPH), NOS uses arginine as a substrate to produce nitric oxide and citrulline. Nitric oxide has a role in normal cellular function, but increased production may be involved in hyperalgesia and may lead to neurotoxicity. |
Although wind-up is very likely an important underlying mechanism contributing to persistent pain, other processes must be considered as contributors to central sensitization. Long-term potentiation (LTP) refers to the strengthening of the efficacy of synaptic transmission that occurs following
activity across that synapse and is a phenomenon that has long been associated with the hippocampus and memory processes (95). Evidence now suggests that an LTP-like phenomenon may also occur within the dorsal horn (96). In contrast to wind-up, which quickly expires following cessation of the stimulus, LTP may be maintained for hours and possible weeks to months following initiation (96,97). Although some have suggested that LTP only occurs under nonphysiologic conditions and therefore has little clinical relevance, the recent demonstration of a pain amplifier within the spinal cord that is switched on by asynchronous, irregular low-frequency inputs in C-fibers supports the clinical relevance of these findings (98). These changes that occur at a cellular level contribute to the development of behavioral alterations that appear to indicate the presence of central sensitization (38). First, there occurs an expansion in receptive field size (99,100), so that a spinal neuron will respond to stimuli that would normally be outside the region that responds to nociceptive stimuli. Second, there occurs an increase in the magnitude and duration of the response to stimuli that are above threshold in strength (101). Last, there occurs a reduction in threshold, so that stimuli that are not normally noxious activate neurons that normally transmit nociceptive information. These changes may be important both in acute pain states such as postoperative pain and in the development of chronic pain.
activity across that synapse and is a phenomenon that has long been associated with the hippocampus and memory processes (95). Evidence now suggests that an LTP-like phenomenon may also occur within the dorsal horn (96). In contrast to wind-up, which quickly expires following cessation of the stimulus, LTP may be maintained for hours and possible weeks to months following initiation (96,97). Although some have suggested that LTP only occurs under nonphysiologic conditions and therefore has little clinical relevance, the recent demonstration of a pain amplifier within the spinal cord that is switched on by asynchronous, irregular low-frequency inputs in C-fibers supports the clinical relevance of these findings (98). These changes that occur at a cellular level contribute to the development of behavioral alterations that appear to indicate the presence of central sensitization (38). First, there occurs an expansion in receptive field size (99,100), so that a spinal neuron will respond to stimuli that would normally be outside the region that responds to nociceptive stimuli. Second, there occurs an increase in the magnitude and duration of the response to stimuli that are above threshold in strength (101). Last, there occurs a reduction in threshold, so that stimuli that are not normally noxious activate neurons that normally transmit nociceptive information. These changes may be important both in acute pain states such as postoperative pain and in the development of chronic pain.
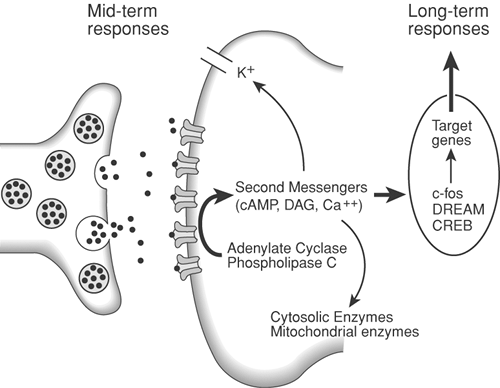 Figure 31-15. Neurotransmitter release from the central terminal of peripheral afferents results in activation of receptor sites on the postsynaptic membrane. Activation of phospholipase C and adenylate cyclase leads to the production of the second messengers cyclic adenosine monophosphate (cAMP) and diacylglycerol (DAG). Mobilization of these second messengers may result in a decrease in potassium (K+) efflux and elevation of intracellular calcium. The increase in intracellular calcium results in the induction of the protooncogene c-fos, production of Fos protein, and a presumed action on target genes to alter long-term responses of the cell to further stimuli. DREAM, downstream regulatory element antagonist modulator; CREB, cyclic adenosine monophosphate (cAMP) response element-binding protein.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





