Reprinted with permission Adam HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke. Stroke. 2007;38:1655–1711.
CT, computed tomography; rtPA, recombinant tissue plasminogen activator.
Chapter Highlights
 The best way to improve neurologic recovery from ischemia is to reduce the degree or the duration of the ischemia.
The best way to improve neurologic recovery from ischemia is to reduce the degree or the duration of the ischemia.
 Ischemia leads to acidosis, glutamate-induced neurotoxicity, sodium (Na) and calcium (Ca) influx, potassium (K) efflux, depolarization, loss of energy production, and free-radical formation. These changes in neurons cause cell death by either necrosis (severe ischemia) or apoptosis (less severe ischemia).
Ischemia leads to acidosis, glutamate-induced neurotoxicity, sodium (Na) and calcium (Ca) influx, potassium (K) efflux, depolarization, loss of energy production, and free-radical formation. These changes in neurons cause cell death by either necrosis (severe ischemia) or apoptosis (less severe ischemia).
 A focal ischemic lesion is induced by the obstruction of blood flow through a cerebral artery. This leads to two areas of insult: the core that is severely ischemic and the penumbra that receives some collateral blood flow and is less ischemic. Neurons die quickly in the core, mainly by necrosis; death in the penumbra is slower and is mainly by apoptosis. Reperfusion and drug therapy are targeted at reducing death in the penumbra of an ischemic lesion.
A focal ischemic lesion is induced by the obstruction of blood flow through a cerebral artery. This leads to two areas of insult: the core that is severely ischemic and the penumbra that receives some collateral blood flow and is less ischemic. Neurons die quickly in the core, mainly by necrosis; death in the penumbra is slower and is mainly by apoptosis. Reperfusion and drug therapy are targeted at reducing death in the penumbra of an ischemic lesion.
 rtPA has been demonstrated to improve outcome from ischemic stroke if administered within 3 hours of stroke onset. In certain patients, this has been extended to within 4.5 hours of onset. rtPA will exacerbate hemorrhagic stroke significantly and lead to uncontrolled bleeding in trauma patients. (See Table 4.1 for contraindications.)
rtPA has been demonstrated to improve outcome from ischemic stroke if administered within 3 hours of stroke onset. In certain patients, this has been extended to within 4.5 hours of onset. rtPA will exacerbate hemorrhagic stroke significantly and lead to uncontrolled bleeding in trauma patients. (See Table 4.1 for contraindications.)
 Maintaining physiologic parameters in the normal range can dramatically reduce ischemia and the damage it causes. It is important to avoid hyperthermia, hyperglycemia, hypotension, hypoxia, hypercapnia, and acidosis. Seizures must be rapidly and effectively treated as they clearly worsen outcome. High intracranial pressure (ICP) should also be avoided and treated if present.
Maintaining physiologic parameters in the normal range can dramatically reduce ischemia and the damage it causes. It is important to avoid hyperthermia, hyperglycemia, hypotension, hypoxia, hypercapnia, and acidosis. Seizures must be rapidly and effectively treated as they clearly worsen outcome. High intracranial pressure (ICP) should also be avoided and treated if present.
 Mild hypothermia (32°C to 35°C) is recommended after cardiac arrest or neonatal asphyxia.
Mild hypothermia (32°C to 35°C) is recommended after cardiac arrest or neonatal asphyxia.
 The effectiveness of drugs for improving clinical outcome from ischemia has not been demonstrated. Barbiturates (thiopental) appear to be most beneficial although the clinical evidence is somewhat equivocal. Some animal evidence exists for beneficial effects of propofol and dexmedetomidine. Volatile anesthetics (isoflurane, sevoflurane, and desflurane) induce preconditioning protection in animals and this may be of benefit to patients. Xenon, erythropoietin, lidocaine, and magnesium have been effective in animal studies and are currently undergoing clinical trials.
The effectiveness of drugs for improving clinical outcome from ischemia has not been demonstrated. Barbiturates (thiopental) appear to be most beneficial although the clinical evidence is somewhat equivocal. Some animal evidence exists for beneficial effects of propofol and dexmedetomidine. Volatile anesthetics (isoflurane, sevoflurane, and desflurane) induce preconditioning protection in animals and this may be of benefit to patients. Xenon, erythropoietin, lidocaine, and magnesium have been effective in animal studies and are currently undergoing clinical trials.
I. IMPORTANT QUESTIONS CONCERNING CEREBRAL PROTECTION
Neurosurgical, cardiac, and vascular procedures carry considerable risk of ischemic/hypoxic injury to the brain and may subject patients to perioperative stroke, long-term adverse neurocognitive outcome, or death.
A. Outcome improvement. In this chapter, we will discuss a number of drugs and measures that may provide cerebral protection. The fundamental question is do they improve clinical outcome?
1. It is important to note that the only treatment proven to improve outcome after ischemic stroke is reperfusion with rtPA within 3 hours of the onset of symptoms.
2. Hypothermia after cardiac arrest and neonatal birth asphyxia has been promising.
3. Most clinical drug trials have failed to improve outcome.
4. Preventing the occurrence of ischemia and reducing the duration and the extent of the ischemia have been the most productive clinical strategies.
B. Importance of mechanisms. In order to understand what may protect against and improve recovery from ischemic brain damage, it is first important to understand the cellular and the molecular mechanisms that cause neuronal damage from ischemic insults.
II. THE ISCHEMIC BRAIN
A. Cerebral ischemia is defined as perfusion insufficient to provide the supply of oxygen and nutrients needed for maintenance of neuronal integrity (which utilizes 40% to 45% of the total cerebral metabolic rate for oxygen consumption [CMRO2]) and neuronal function (which utilizes 55% to 60% of CMRO2).
1. Cellular integrity involves basic cellular activities such as carbohydrate, lipid, and protein synthesis and the maintenance of ionic gradients and membrane integrity.
2. Neuronal function involves the generation and the transmission of nerve impulses and is manifested by the presence of electroencephalographic (EEG) activity.
3. Ischemia leads to the depletion of adenosine triphosphate (ATP), which is required for all active cellular processes. There is a hierarchy of ischemic damage in which neuronal function is abolished before cellular integrity is lost.
B. Types of cerebral ischemia
1. Global cerebral ischemia
a. Complete global ischemia is the complete cessation of blood flow to the brain as is seen with cardiac arrest. Neuronal depolarization occurs in a few minutes. The hippocampus and the cerebral cortex are most vulnerable and the neurons in these regions die rapidly.
b. Incomplete global ischemia occurs with hypotension or shock. The level of cerebral blood flow (CBF) is less than that required for normal function.
2. Focal cerebral ischemia involves the occlusion of a single cerebral artery leading to ischemia that is confined to a specific region in the brain.
a. The ischemic region is composed of a severe ischemic area, the core, which is surrounded by a less severe ischemic area, the penumbra.
b. The neurons in the core die rapidly whereas those in the penumbra can survive for a longer period of time and can be rescued from infarction if recanalization (i.e., reperfusion) occurs early. This provides a window of opportunity for therapeutic intervention.
c. If the blood flow is not restored, however, the infarct core will grow progressively and the neurons in the penumbra will also die.
C. The ischemic cascade (Fig. 4.1). The ischemic insult evokes a cascade of pathophysiologic processes involving a multitude of chemical mediators of neuronal damage.
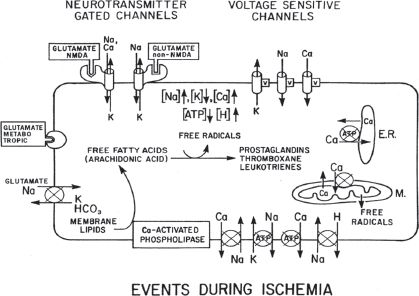
FIGURE 4.1 Effect of ischemia on ion and metabolite levels in neurons. For clarity, ion channels are shown on the top membrane and ion pumps on the bottom membrane; their actual location can be on any membrane surface. Circles indicate energy-driven pumps; an x through the circle indicates that this pump is blocked or has reduced activity during ischemia. V indicates a voltage-dependent channel. NMDA, N-methyl-D-aspartic acid; ATP, adenosine triphosphate. (From Bendo AA, Kass IS, Hartung J, et al. Anesthesia for neurosurgery. In: Barash PG, Cullen BF, Stoelting RK, eds. Clinical Anesthesia, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:746–789, with permission.)
1. The decreased availability of oxygen and glucose results in depletion of ATP. This depletion occurs within 2 to 4 minutes of the onset of complete ischemia.
a. Phosphocreatine (PCr) is a source of high-energy phosphate that allows the resynthesis of ATP from adenosine diphosphate (ADP). Brain PCr levels are normally three times those of ATP.
b. PCr levels decrease first with the onset of ischemia and act as a buffer to maintain ATP levels. Once PCr levels fall, ATP levels diminish rapidly thereafter.
2. Lactate levels increase as a result of the anaerobic metabolism of glucose. Lactic acidosis aggravates ischemic damage and promotes free-radical formation. This is followed by the lipid peroxidation of cell membranes.
a. The continued supply of glucose associated with incomplete ischemia contributes to anaerobic glycolysis. Lactic acid production is increased, which is thought to be the mechanism for the heightened damage seen with incomplete ischemia.
b. In contrast, complete ischemia results in the complete cessation of glycolysis and no additional lactic acid production once all the glucose and the glycogen have been depleted.
c. Hyperglycemia is an independent risk factor for exacerbating ischemic damage. The primary mechanism for the increased damage appears to be the enhanced production of lactate and intracellular acidosis.
3. Cerebral ischemia increases the release of the excitatory amino acid (EAA) neurotransmitters glutamate and aspartate.
a. Three receptors for the excitatory neurotransmitters are currently identified.
1. N-methyl-D-aspartate (NMDA) receptors mediate the influx of Na and Ca and the efflux of K through receptor-coupled ion channels. These channels are important for long-term potentiation, learning, and memory. The presence of glycine is necessary for activation of NMDA receptors by glutamate. Magnesium (Mg) blocks the channel at its physiologic concentration. This block can be overcome, however, by neuronal depolarization. The NMDA channels open during hypoxia and ischemia. This leads to a large influx of Na and Ca, which contributes to neuronal damage.
2. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors mediate the influx of Na and the efflux of K. A small percentage of these channels have a subunit composition that can also allow Ca to pass through them. These channels contribute to damage by allowing Na into the cell, which when the water comes with it, causes cell swelling and death and also leads to depolarization, which facilitates the activation of the NMDA channels.
3. Kainate receptors also mediate the influx of Na and the efflux of K.
b. Glutamate stimulates all three receptors, but aspartate affects only the NMDA receptor.
c. Glutamate causes neuronal cell death from immediate and delayed neurotoxicity.
1. In immediate neurotoxicity, glutamate activates the NMDA receptor, leading to the influx of Ca, Na, chloride (Cl), and water (H2O), which results in cellular edema, membrane lysis, and cell death.
2. In delayed neurotoxicity (24 to 72 hours), the activated NMDA receptor promotes a cycle of damage initiated by the influx of Ca. This leads to the activation of phospholipases and proteases, the formation of free radicals and free fatty acids (FFAs) including arachidonic acid, and cell death.
4. The increased cytosolic Ca concentration is an important event in the ischemic cascade leading to cell death and is caused by several mechanisms.
a. Depletion of ATP results in failure of the energy-requiring Na/K ATPase-dependent ion pumps. Na and Cl build up intracellularly and K extracellularly. Influx of H2O and edema occur secondary to the intracellular ion level increase. The resulting membrane depolarization leads to opening of voltage-sensitive Ca channels and Ca influx.
b. The increase in intracellular Na leads to the loss of the Na gradient, which is an energy source for the Na/Ca exchanger that pumps Ca out of the cell.
c. Depletion of ATP results in the failure of the plasma membrane Ca ATPase pump and reduced pumping of Ca out of the cell.
d. Decreased ATP leads to reduced activity of the Ca ATPase pump in the endoplasmic reticulum (ER), which removes Ca from the cytosol and pumps it into the ER. The reduced pumping leads to a net loss of Ca from the ER.
e. EAA levels increase during ischemia, leading to stimulation of glutamate receptors and the opening of NMDA-mediated Ca channels.
5. Numerous effects of high cytosolic Ca form a common pathway leading to neuronal cell destruction.
a. Increased intracellular Ca activates phospholipases A1, A2, and C, which lead to the hydrolysis of membrane phospholipids and the release of FFAs. The loss of membrane phospholipids also results in mitochondrial and cell membrane damage.
1. Arachidonic acid, a major FFA, is a precursor to prostaglandins, leukotrienes, and free radicals.
2. Both prostaglandins (via the cyclooxygenase pathway) and leukotrienes (via lipoxygenase) lead to cerebral edema.
3. Thromboxane A2, a prostaglandin derived from arachidonic acid with potent vasoconstrictor and platelet aggregation properties, potentiates ischemia and has been implicated in reperfusion injury.
4. Free radicals cause lipid peroxidation within neuronal cell membranes. This alters membrane function and damages cellular membranes.
b. Ca-activated proteases called calpains break down essential proteins and lead to damage.
c. Ca influx into mitochondria during ischemia can lead to mitochondrial dysfunction and damage.
6. Free-radical formation
a. Free radicals can be formed independent of Ca activation by incomplete oxidation in the mitochondria or by arachidonic acid metabolism after ischemia and acidosis.
b. Free radicals damage proteins and membranes and inhibit their function.
7. Activation of cell death pathways: necrosis versus apoptosis
a. Apoptosis is orderly programmed cell death that requires energy to be carried out. It can be modulated and reversed by trophic factors (Fig. 4.2) and involves:
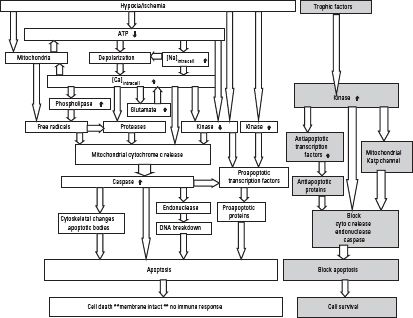
FIGURE 4.2 Ischemic cascade leading to apoptosis. ATP, adenosine triphosphate. (From Kass IS. Brain metabolism and mechanisms of cerebral ischemia. In: Schwartz A, Butterworth J, Gross J, eds. Refresher Courses in Anesthesiology. Philadelphia, PA: Lippincott Williams & Wilkins, 2006;34:85–93.)
1. Milder ischemia
2. Mitochondria cytochrome C release and activation of caspase and endonuclease
3. Compartmentalization of cell contents (apoptosomes)
4. Reduced exposure of cell contents to outside, which limits inflammation
5. Primary form of cell death in the penumbra of a region of focal ischemia
b. Necrosis leads to cell disintegration and involves:
1. More severe ischemia
2. Cell and mitochondria swelling and rupture of lysosomes
3. Cell lysis and bursting
4. Exposure of intracellular contents, which causes inflammation and damage to surrounding cells
5. Primary form of cell death in the core of an ischemic region
D. Clinical aspects of ischemia
1. Of all body organs, the brain is the most vulnerable to ischemia. Loss of consciousness occurs within 15 seconds of cardiac arrest. Brain PCr levels fall to very low levels within 1 minute. Glucose and ATP stores are exhausted within 4 to 5 minutes. Critical levels for CBF, cerebral perfusion pressure (CPP, the difference between the mean arterial pressure [MAP] and the intracranial pressure [ICP]), and the partial pressure of arterial oxygen (PaO2) have been determined below which cerebral ischemia occurs with characteristic EEG changes. With prolongation of the cerebral ischemia, progression to isoelectricity in the EEG occurs quickly.
2. Critical CBF is 18 to 20 mL/100 g of brain/minute. The EEG becomes isoelectric at a CBF of 15 mL/100 g/minute. Metabolic failure occurs at a CBF of 10 mL/100 g/minute. The penumbra is a hypoperfused region that may remain viable for a period of time after the insult occurs.
3. Critical CPP is 50 mm Hg in the normal individual.
4. Critical PaO2 is 30 to 35 mm Hg in healthy awake patients.
E. Reperfusion injury refers to damage that occurs after the restoration of cerebral perfusion.
1. This can be due to processes activated during ischemia that require energy or oxygen for their culmination.
a. Free radicals are increased in the period immediately after ischemia.
b. Apoptosis requires energy-using processes to be completed. In the absence of energy, these cells die of necrosis.
2. Reperfusion injury can also be due in part to the inflammatory response of damaged tissue. In response to tissue damage, white blood cells, carried to the area by the newly restored blood flow, release inflammatory mediators as well as free radicals, which may exacerbate the neuronal damage.
III. PATIENTS WHO WOULD BENEFIT FROM CEREBRAL PROTECTION
A. Patients who require resuscitation from cerebral ischemic damage include:
1. Intensive care unit (ICU) patients who have traumatic but non-operative brain injury such as diffuse axonal injury (DAI) with increased ICP and cerebral edema who may be candidates for barbiturate coma.
2. Patients who have Reye′s syndrome and cerebral edema with increased ICP.
3. Near-drowning victims who have anoxic encephalopathy, cerebral edema, and intracranial hypertension.
4. Stroke patients who have non-hemorrhagic stroke who may be candidates for fibrinolytic therapy with rtPA (see Section IV-C).
5. Patients who have had a cardiac arrest with re-establishment of circulation.
6. Patients who have neonatal birth asphyxia.
B. Candidates for cerebral protection also include patients who are at risk for cerebral ischemia/reperfusion injury:
1. Patients who have an increase in intracranial volume from tumor, abscess, hematoma, hydrocephalus, or chronic cystic fluid collections with or without increased ICP who are scheduled for neurosurgical procedures.
2. Patients who are scheduled for intracranial vascular procedures, such as cerebral aneurysm coiling/clipping, excision of arteriovenous malformation (AVM) and cavernous angioma, and extracranial vascular procedures including carotid endarterectomy (CEA) and superficial temporal artery to middle cerebral artery (STA-MCA) bypass, which involve temporary vessel occlusion and the possibility of focal ischemia.
3. Patients who are scheduled for the clipping or coiling of giant or complex basilar artery aneurysms.
4. Cardiopulmonary bypass (CPB) patients who are at risk for either global cerebral ischemia from low-flow states or focal cerebral ischemia from multiple small emboli.
C. Patients who are at risk for cardioembolic strokes
Cardioembolism accounts for about 20% of all ischemic strokes. Cardioembolic strokes have a high recurrence rate and are often preventable, so that primary and secondary prevention strategies are important.
1. Clinical features of cardioembolic stroke may be helpful in distinguishing cardioembolism from other causes of cerebral ischemia, but they are not very sensitive. These include short time from sudden onset to maximal deficit, rapid recovery from symptoms, simultaneous or sequential strokes in different cerebral arterial territories, and onset of symptoms after a Valsalva-provoking activity.
2. Antiplatelet and anticoagulant therapies
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


