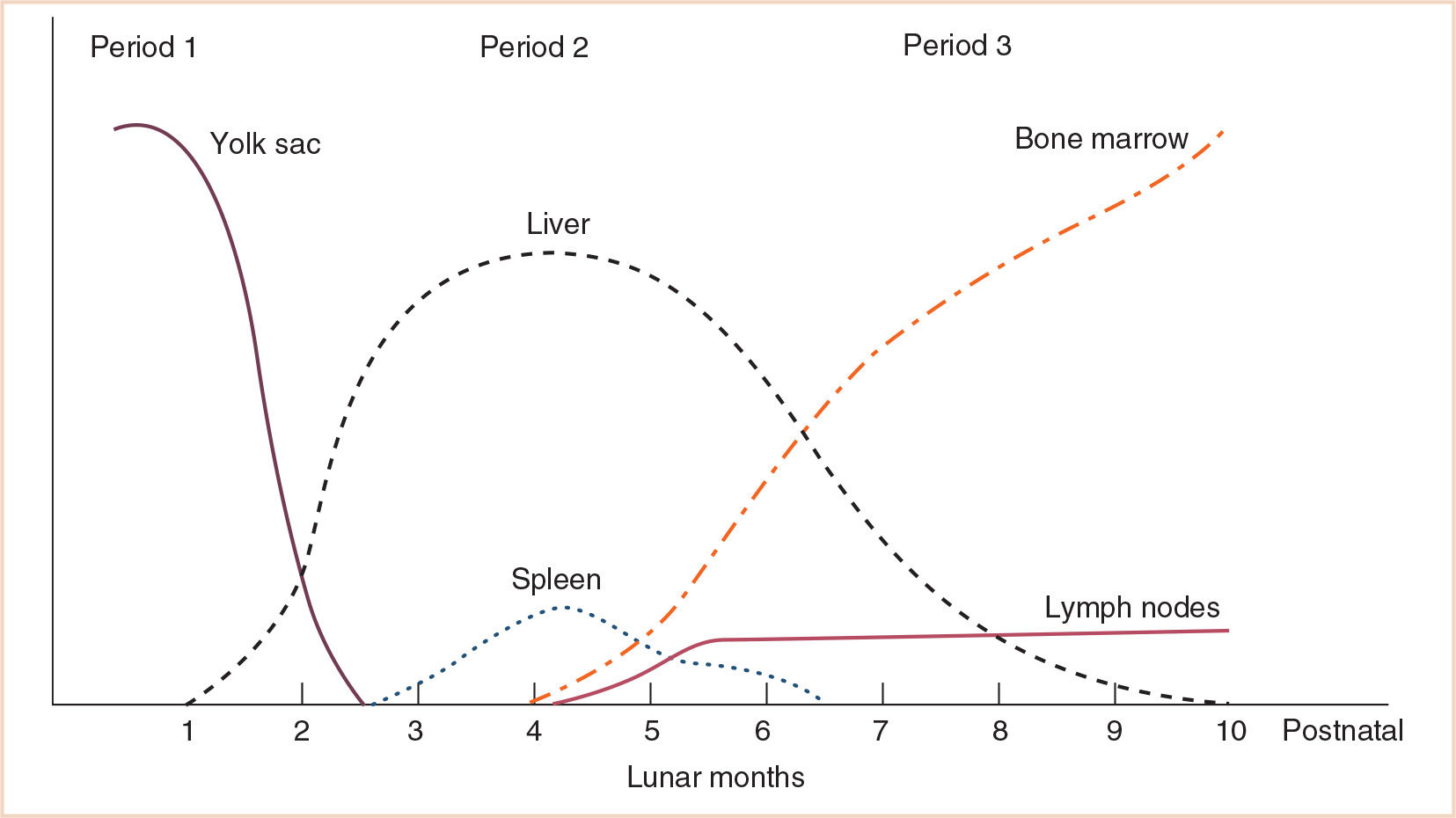
1. During fetal life, hematopoiesis occurs in several distinct locations, with sequential timing of cellular elements produced during gestation.
2. The primary problem in anemia is impaired oxygen-carrying capacity of the blood with resultant impaired oxygen delivery to the body.
3. The presence of anemia can be a bellwether for many other diseases; therefore, in the absence of a primary hematologic disorder, a search for the underlying cause of unexplained anemia should be sought.
4. Patients with sickle cell disease should be carefully managed in the perioperative period to prevent complications such as vaso-occlusive crises, acute chest syndrome, and stroke.
5. Hemophiliacs and patients with von Willebrand disease require careful treatment with desmopressin or factor concentrates in the perioperative period to decrease bleeding complications.
Children born with a number of congenital erythrocyte disorders such as hereditary spherocytosis, sickle cell disease, or one of the thalassemias may have reason to present to the operating room (OR) or diagnostic imaging suite. Some operative procedures such as splenectomy or cholecystectomy are related to an underlying red blood cell (RBC) defect. The development and competence of the immune system relies on the exquisite interaction of cellular and molecular components of leukocyte and lymphocyte lines as well as various immunoglobulins, plasma proteins, cytokines, and chemokines which will be discussed in Chapter 32. The leukemias are often the cause for surgical procedures such as central venous line (CVL) placement, bone marrow aspirate and biopsy, lung biopsy, and exploratory laparotomy. Platelet disorders may be acquired or congenital in association with certain syndromes. Inherited disorders of the coagulation system such as classic hemophilia and other hemophilias to the very common von Willebrand disease (vWD) also impact the perioperative care of children.
I. Embryology of hematopoeisis
A. During fetal life, hematopoiesis occurs in several distinct locations. These locations vary in the cell types they contain throughout gestation. The yolk sac is the first location for hematopoiesis, which occurs from the second through the 6th to 8th weeks of gestation. At around this time, hematopoiesis begins in the liver, where the process continues throughout the balance of gestation. As bone marrow production of blood elements begins during the second trimester, liver hematopoiesis diminishes (1–3).
B. In addition to the different locations for the production of the formed elements of the blood, there are also sequences in the timing of cellular elements produced during gestation. For example, in all hematopoietic locations, macrophages are produced before granulocytes. As gestation progresses, the proportion of macrophages decreases and that of granulocytes increases. The production of erythrocytes, first in the yolk sac, then liver and finally in the bone marrow, gradually increases throughout gestation. The hemoglobin (Hb) concentration increases from approximately 12 g per dL at 20 weeks gestation to 16 to 17 g per dL at term. Figure 28.1 shows the locations of blood cell production during embryonic and fetal development. Figure 28.2 is a schematic of the maturation of various blood cell lines.
II. Abnormalities of erythrocytes
A. Background: The definition of anemia varies with the age of the child. A person whose Hb concentration is below the lower end of the range listed in Table 28.1 is considered anemic. The causes for anemia are generally divided into those that result from decreased production of RBCs and those due to increased destruction of RBCs. Table 28.1 shows the range of normal values for Hb concentration throughout childhood. Table 28.2 lists the common causes for anemia in infancy and childhood (4).
The primary problem in anemia is impaired oxygen-carrying capacity of the blood with resultant impaired oxygen delivery to the body. Physiologic responses to this diminished oxygen-carrying capacity include increased cardiac output (primarily heart rate), increased oxygen extraction, increased RBC 2,3-diphosphoglycerate (2,3-DPG), and greater production of erythropoietin (EPO). As anemia worsens beyond the ability of the compensatory mechanisms, easy fatigability, tachypnea, shortness of breath, and even congestive cardiac decompensation will occur.
The presence of anemia can be a bellwether for many other diseases; therefore, in the absence of a primary hematologic disorder, a search for the underlying cause of unexplained anemia should be sought.
B. Embryology
1. RBC production is controlled only by fetal growth factors.
2. Maternal EPO does not cross the placenta. The liver produces fetal EPO during approximately the first 24 weeks of gestation. In the last trimester and continuing into postnatal life, EPO production moves to the kidney.
3. During fetal and postnatal life, Hb comprises several different types of globin molecules.
a. During the first 10 weeks of development, embryonic Hb is present.
b. There is also a small amount of fetal and adult Hb present.
c. The percentage of fetal Hb (α2–γ2) increases from 20% in the first few weeks of embryonic life to a peak of 80% at 10 to 12 weeks.
d. Adult Hb (α2–β2) makes up approximately 10% of the total Hb from early embryonic life until 20 weeks of gestation then gradually increases to 20% at term.
e. Following birth, the percentage of fetal Hb declines from 80% to approximately 0% by 6 months of age with a corresponding increase in adult (α2–β2) Hb throughout embryonic and fetal development (1–3).
C. Pathophysiology
1. Anemia caused by decreased RBC production is generally seen in infants and young children.
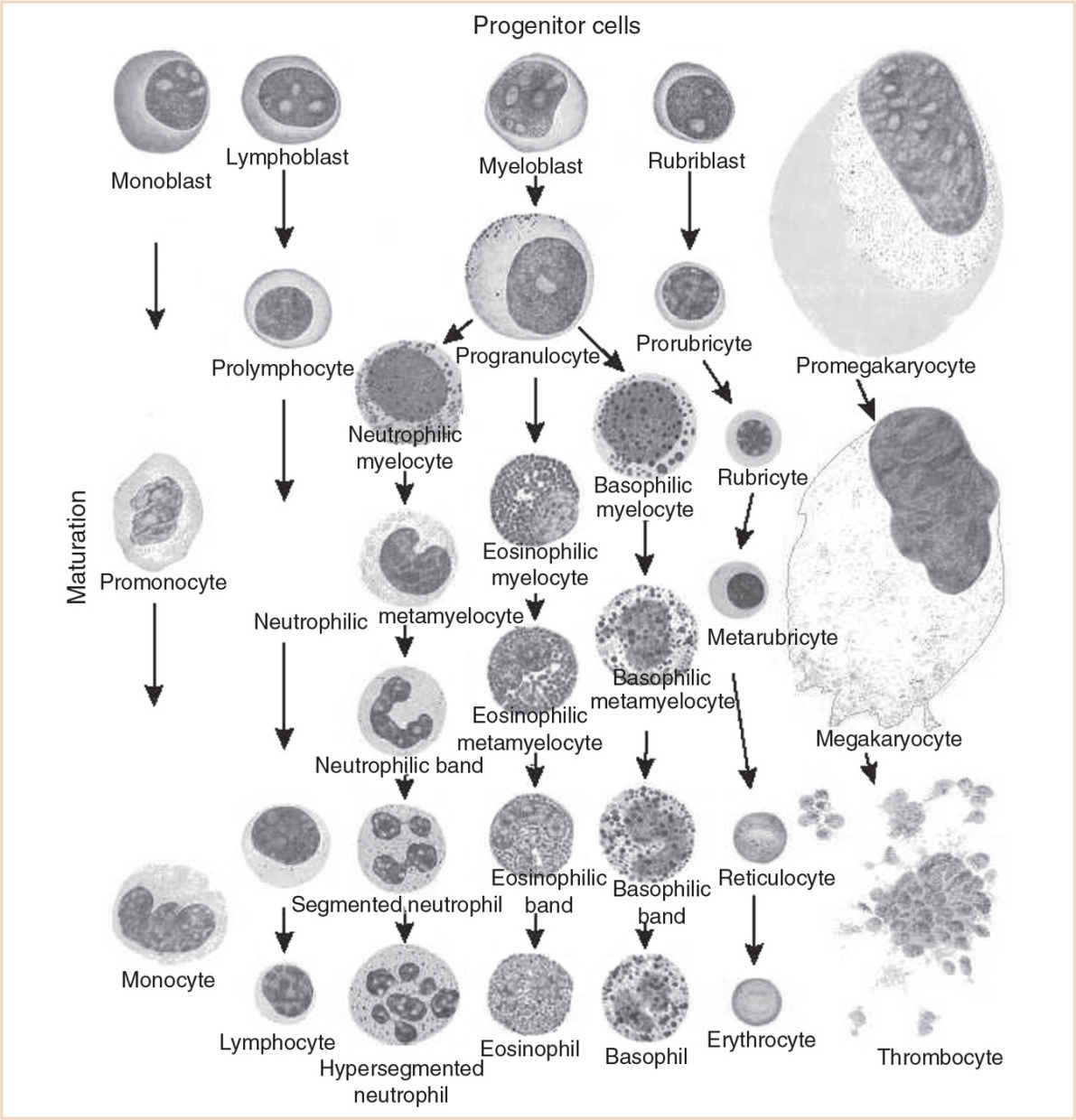
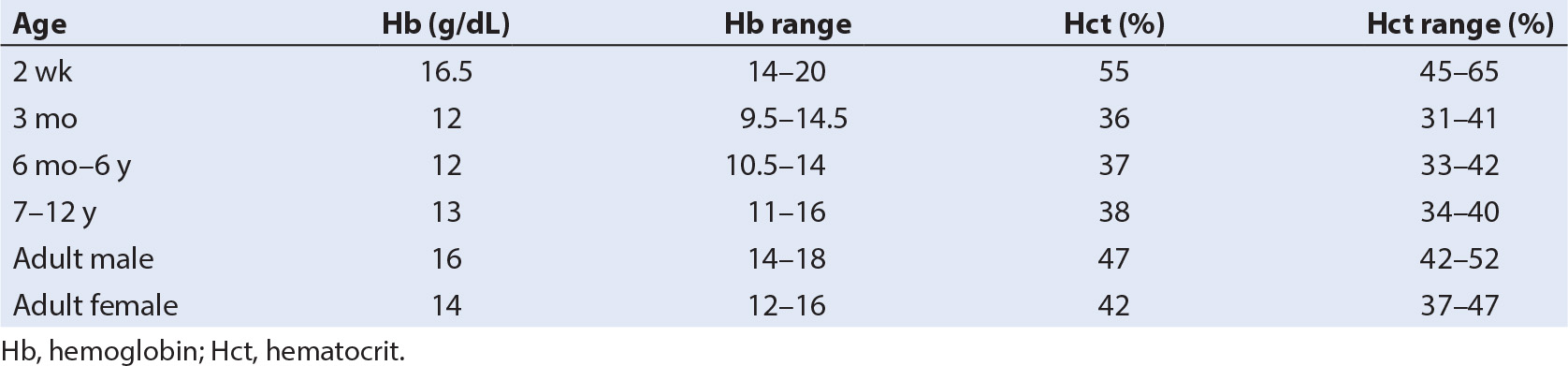
TABLE 28.2 Common causes of anemia in infancy and childhood
Etiologies of anemia due to decreased RBC production
Transient erythroblastopenia of childhood
Diamond–Blackfan anemia
Physiologic anemia of infancy
Iron deficiency anemia
Etiologies of hemolytic anemia
RBC membrane defects: hereditary spherocytosis, hereditary elliptocytosis, and paroxysmal nocturnal hemoglobinuria
RBC enzyme deficiencies: G6Pd deficiency, pyruvate kinase deficiency
Hemoglobinopathies: thalassemia, sickle cell disease
Immunologic: autoimmune hemolytic anemia (AIHA), hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura, hemolytic disease of the newborn
Hypersplenism
Extrinsic damage: DIC, prosthetic heart valves, ECMO, cardiopulmonary bypass
RBC, red blood cell; Hb, hemoglobin; DIC, disseminated intravascular coagulation; ECMO, extracorporeal membrane oxygenation.
a. Transient erythroblastopenia of childhood (TEC) is seen in infants and children.
(1) The onset is usually between 6 months and 3 years of age and often follows a viral illness.
(2) The cause is bone marrow suppression on an immunologic basis.
(3) The peripheral smear shows few to no reticulocytes and the bone marrow shows a deficiency of RBC precursors.
(4) Most affected children have normal white blood cell (WBC) counts and elevated platelet counts. Examination of the bone marrow is generally not needed to confirm the diagnosis, but in specific individuals that test may be done (5).
b. Less common but more serious is congenital hypoplastic anemia, also known as Diamond–Blackfan anemia (DBA).
(1) Most cases of this condition are diagnosed within the first year of life.
(2) The genetic defect involves mutations in various ribosomal proteins and in proteins that process ribosomal RNA.
(3) In these children there are few reticulocytes in the peripheral blood and the bone marrow shows a deficiency of RBC precursors.
(4) Most affected children have normal hematopoiesis in fetal life. Clinical manifestations of severe anemia are seen within the first 6 months of life in many infants (6).
(5) Many children affected with Diamond–Blackfan anemia have skeletal, renal, ocular or craniofacial anomalies, which may have implications for the anesthesiologist.
(6) Treatment includes corticosteroids, transfusion therapy, iron chelation, growth factors, and stem cell transplantation. Some children develop hypersplenism.
(7) Patients have a reduced life expectancy due to treatment side effects and risk of hematologic malignancy (7).
c. Physiologic anemia of infancy is a term that refers to the expected decline in Hb concentration seen in normal newborns.
(1) At term, the range of Hb concentration is 14 to 20 g per dL.
(2) Shortly after birth, the Hb concentration begins to decrease.
(3) This process continues for approximately 2 months with the result that Hb concentration at 3 months of life is 10 to 14 g per dL.
(4) During this period there is also a switch from fetal Hb with its higher affinity for oxygen to lower affinity adult Hb.
(5) In preterm newborns, the Hb concentration at birth is lower and the nadir of the physiologic anemia is lower.
d. Anemia due to iron deficiency can be a problem for infants fed cow’s milk who do not receive iron supplementation (8).
(1) Adolescent females are another at-risk group for iron deficiency anemia.
e. Anemia of chronic disease is an acquired disorder characterized by abnormal iron hemostasis and impaired erythropoiesis (9).
2. Anemia caused by excessive destruction of RBC as opposed to impaired production manifests as hemolytic anemia and can be seen at any age.
a. RBC lifespan is normally 110 to 120 days, but is shorter with hemolysis.
(1) The bone marrow responds to the increased destruction of RBCs with increased production of these blood cells, manifest by increased numbers of reticulocytes in the peripheral smear.
(2) As a result of excessive hemolysis, the products of Hb metabolism are present in increased amounts.
(3) Unconjugated bilirubin concentrations in the serum are elevated and calcium bilirubinate gallstones may be formed.
(4) If splenomegaly is noted in the context of hemolytic anemia, splenectomy may be indicated.
(5) Treatment varies from folate supplementation to transfusion to splenectomy depending on the severity and type of anemia.
b. The hereditary membrane defects (hereditary spherocytosis and elliptocytosis) can be diagnosed based on RBC morphology seen on a peripheral blood smear. Abnormalities of the RBC membrane lead to decreased surface area, decreased deformability and increased fragility, resulting in splenic sequestration and destruction of RBCs (10).
c. If the RBCs are not exposed to oxidative stress (i.e., infection, medications), G6PD deficiency-related hemolysis can be minimized.
d. Hemolytic disease of the newborn occurs from alloimmunization of RBC antigens from circulating maternal antibodies crossing the placenta. Severity of disease can be variable, from mild jaundice to hydrops fetalis with treatment ranging from phototherapy to exchange transfusion.
e. Autoimmune hemolytic anemia (AIHA) is a rare disorder caused by autoantibodies (i.e., warm-reactive, cold-agglutinin) to RBC antigens. Diagnosis is with direct antiglobulin or Coombs’ test. Treatment includes immunosuppressives and/or plasmapheresis (11).
f. The defect in the thalassemias consists of an imbalance in the globin chains that make up Hb. In β-thalassemias, there is an excess of α globin chains while the γ and δ chains are produced in normal numbers. This leads to tetramers of four α chains, or two α and two γ (Table 28.3). In α-thalassemias, there is an excess of β-globin chains. The excess globin chains interact with the red cell membrane, causing oxidative injury and premature RBC death.
g. In sickle cell anemia, the deoxygenated Hb molecules distort the cell membrane, producing a sickle shape, as a result of aggregation along the longitudinal axis of the cell. These sickled cells become dehydrated and rigid and cause tissue infarcts by impeding blood flow and oxygen delivery to tissues. Patients heterozygous for HbS and another abnormal Hb such as HbC (Hb of thalassemia) may also have end-organ disease and signs and symptoms of organ infarction. These patients have risks similar to those for patients with sickle cell disease (HbSS).
CLINICAL PEARL Children with SS disease and other sickle hemoglobinopathies such as SC disease or sickle thalassemia undergo procedures that put them at significant risk for acute chest syndrome in the post op period including cholecystectomy or splenectomy.
TABLE 28.3 The thalassemias
| Presentation | Consequence |
Beta | ||
Homozygous β-thalassemia (Cooley’s anemia; thalassemia major) | Severe, progressive hemolytic anemia in the second to sixth months of life | Chronic transfusion requirement; hypertrophy of extramedullary hematopoietic tissue; pallor; hemosiderosis; jaundice; hypersplenism; hepatomegaly; developmental delay; growth failure; diabetes; cardiac failure |
Homozygous β+ thalassemia (thalassemia intermedia) | Milder form, also known as Cooley’s-anemia-like syndrome |
|
Alpha |
|
|
Alpha silent carrier—deletion of 1/4 α-globin genes | Mild microcytosis; present in 25% of black Americans | None to minimal symptoms |
Alpha trait—deletion of 2/4 α-globin genes | Mild microcytic anemia |
|
Hb H disease—deletion of 3/4 α-globin genes | Thalassemia-intermedia-like syndrome; anemia and abnormal RBC morphology | Transfusion support in the setting of metabolic or oxidative stressors |
Alpha-hydrops fetalis—deletion of 4/4 α-globin genes | Total absence of α-chain synthesis; infants are severely hypoxic because of high oxygen affinity of Hb species present (Hb Bart; no HbA or HbF). | Usually stillborn or neonatal death |
Hb, hemoglobin; RBC, red blood cell.
III. Physiologic considerations for specific anemias
A. Transient erythroblastopenia of infancy: Considerations for patients treated with steroids (4,5).
B. Hereditary spherocytosis/elliptocytosis: Increased fragility and destruction of RBCs. Can range from asymptomatic carrier state to severe, chronically transfusion-dependent state. Splenomegaly with sequestration, gallbladder disease, hemolytic crisis associated with acute viral illness (10).
C. G6PD deficiency: Medications causing hemolysis include sulfa drugs, quinidine, prilocaine, lidocaine, antimalarial drugs, antipyretic drugs, nonnarcotic analgesics, vitamin-K analogs, and (maybe) sodium nitroprusside.
D. Thalassemias: Infection, acute chest syndrome, stroke, splenomegaly with splenic sequestration, renal insufficiency, gallbladder disease, endocrinopathies, skin ulcers, extramedullary hematopoiesis leading to pathologic fractures, and alterations in facial features with occasional reports of airway difficulty (12,13).
E. Sickle cell disease: Pain crises, infection, acute chest syndrome, stroke, splenic sequestration, renal insufficiency, gallbladder disease, endocrinopathies and skin ulcers. Pathological end-organ damage in sickle cell states is due to the following (14):
1. Sickling of cells in blood vessels causing infarcts and subsequent tissue destruction due to ischemia.
2. Hemolytic crises secondary to hemolysis.
3. Aplastic crises that occur with bone marrow exhaustion (parvovirus B19).
IV. Anesthetic considerations for specific anemias
A. Hereditary RBC membrane defects
1. Possible hemolysis from alterations in osmolality or temperature.
2. Effects of chronic transfusion and iron overload such as cardiomyopathy, liver fibrosis, pancreatic β-cell fibrosis with development of diabetes mellitus, and side effects from iron chelation with deferoxamine such as hearing loss.
3. Immunocompromised state and increased risk of sepsis from encapsulated organisms after splenectomy.
B. RBC enzymatic deficiencies
1. Avoid certain medications in patients with G6PD deficiency.
2. No specific considerations for patients with pyruvate kinase deficiency.
C. Abnormal hemoglobins
1. Effects of hemolysis, chronic transfusion and iron overload such as cardiomyopathy, liver fibrosis, pancreatic β-cell fibrosis with development of diabetes mellitus, and side effects from iron chelation with deferoxamine such as hearing loss, effects of hematopoietic stem cell transplantation (HCST) for HbSS.
2. Immunocompromised state and increased risk of sepsis from encapsulated organisms after splenectomy.
3. Thalassemias: Careful evaluation of severity of disease with degree of end-organ involvement.
4. Sickle cell disease (15–24)
a. Preoperative evaluation: Eight percent to 10% of black Americans have sickle cell trait (HbAS); 0.2% are homozygous (HbSS) (see Table 28.4). HbAS is not a clinically significant disorder for anesthesia because these cells begin to sickle only when oxygen saturation is below 20%.
(1) History of prior vaso-occlusive crises
(2) History of neurologic damage or prior strokes
(3) Prior acute chest syndrome episodes
(4) History of cardiac failure, myocardial infarction
(5) History of renal dysfunction
(6) Preprocedure analgesic, nonsteroidal anti-inflammatory drug (NSAID), and/or opioid use
b. Preoperative transfusion: Consider the specific patient’s general condition, the starting Hb, the percentage of HbS, and the nature of the surgical procedure. Indications for RBC exchange transfusion include acute chest and stroke. Exchange transfusion (target—increase HbA to 40% and Hct to 35%) may be beneficial because of the following:
(1) Improved clinical symptoms
(2) Improved blood oxygenation
(3) Decreased serum bilirubin levels
(4) Improvement of pneumococcal meningitis
(5) Decreased iron load
(6) Cessation of hematuria in papillary necrosis
The risks of exchange transfusion, however, may exceed the benefit!
(1) Exposure to more blood units
(2) Increased venous access requirements
Recent studies suggest a single preoperative transfusion to a total Hb concentration of 10 g/dL is as effective as an aggressive regimen to decrease the HbS to 30% or less in preventing postoperative complications (17).
TABLE 28.4 Common comorbidities in sickle cell syndromes
Pulmonary dysfunction with increased shunt
Renal insufficiency
Gallstones
Myocardial infarction
Priapism
Stroke
Aseptic necrosis of bones and joints
Ischemic ulcers
Retinal detachment from neovascularization
Complications of repeated transfusions
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


