Hematology Disorders
Somasundaram Jayabose MD
INTRODUCTION
Anemia is defined as a reduction in the hemoglobin or hematocrit values below the lower limit of normal for age (see Table 42-1). Possible causes include blood loss, increased destruction of red cells (hemolysis), and decreased production of hemoglobin or red cells. In most cases, it is possible to determine the cause of anemia with a methodical approach to diagnosis. Iron deficiency anemia, thalassemia minor, and anemia of infection or inflammation are among the most common anemias in children. In the vast majority of cases, primary care providers can diagnose and manage these conditions. A consultation with a pediatric hematologist is necessary when anemia is severe (hemoglobin less than 8 gms/dL) or when there is associated thrombocytopenia, neutropenia, or both. Children with inherited anemia require regular visits with both the primary care provider and the pediatric hematologist.
 |
A careful history and physical exam can offer several clues to the anemia’s cause (see Table 42-2 and Table 42-3). By evaluating a few initial laboratory data (ie, mean corpuscular volume (MCV), red cell distribution width (RDW), reticulocyte count, blood smear findings, serum lactic dehydrogenase (LDH), and indirect bilirubin level), providers can classify most anemias into one of the following five groups:
 |
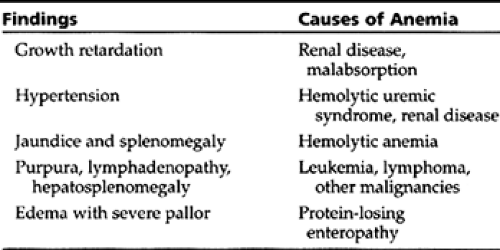 |
Microcytic anemias
Normocytic anemias
Macrocytic anemias
Hemolytic anemias
Anemias caused by bone marrow failure
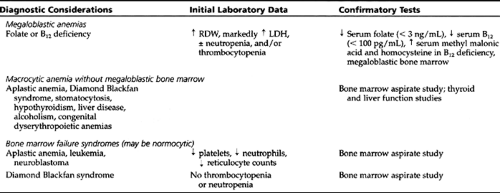
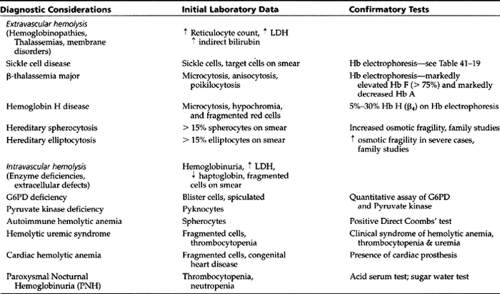
The differential diagnosis for each group and the necessary confirmatory tests are given in Table 42-4, Table 42-5, Table 42-6, and Table 42-7. The differential diagnosis of neonatal anemia is provided in Table 42-8.
 |
 |
 |
 |
 |
• Clinical Pearl
An easy way to remember the lower limit of normal MCV is to add the child’s age in years to the number 70.
Anemia is considered microcytic when the MCV is less than the lower limit of normal for age (see Table 42-1). Microcytic anemia, when mild (hemoglobin not less than 8 gms/dL) and not associated with any systemic illness, is most often secondary to iron deficiency anemia (IDA) or thalassemia minor (trait).
• Clinical Pearl
In an anemic child, a low MCV with a high RDW usually is associated with iron deficiency.
Even more specifically, an RDW index (MCV/RBC × RDW) greater than or equal to 220 supports the diagnosis of IDA with more than 90% sensitivity and specificity (Jayabose, 1999). Such a patient can be empirically started on iron therapy. A response to iron therapy (an increase in the hemoglobin level of at least 1 gm/dL in 2 weeks and to normal values within 4 to 6 weeks) confirms the diagnosis. An algorithm for the diagnosis of mild microcytic anemia is given in Figure 42-1.
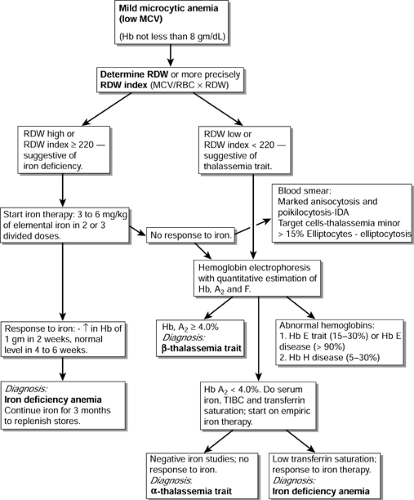 Figure 42-1 Algorithm for the diagnosis of mild microcytic anemias. |
• Clinical Pearl
Patients with microcytosis and a normal or low RDW, or those who fail empiric iron therapy, may have thalassemia minor and should be evaluated by a hemoglobin electrophoresis. An RDW index (MCV/RBC X RDW) less than 220 is strongly suggestive as well.
• Clinical Pearl
A hemoglobin A2 level greater than 4.0% is diagnostic of β-thalassemia trait.
Hemoglobin electrophoresis is also diagnostic in cases of Hemoglobin E disease, which is most common in persons of Southeast Asian descent. Presence of microcytic anemia in one or both parents supports this diagnosis. Parents and siblings of a child diagnosed with thalassemia trait (α or β) should be evaluated by a complete blood count (CBC), and those who have microcytic anemia should be evaluated by hemoglobin electrophoresis. If both parents have thalassemia trait, they should receive formal genetic counseling because of the risk of thalassemia major in future progeny.
• Clinical Pearl
A normal hemoglobin electrophoresis (with normal Hb A2 and Hb F) in a patient with microcytic anemia not responding to iron therapy is very suggestive of alpha thalassemia trait.
This chapter discusses in detail the various types of anemia. It also provides information about the various neutropenias. Finally, it examines different bleeding disorders.
MICROCYTIC ANEMIAS
Iron Deficiency Anemia (IDA)
Iron deficiency anemia (IDA) occurs because of inadequate intake of iron or blood loss. IDA from inadequate intake occurs most commonly between ages 9 months to 24 months. To maintain positive iron balance during childhood, the body must absorb 0.8 mg of elemental iron each day from the diet. Because the body absorbs less than 10% of dietary iron, the daily diet should contain at least 10 mg of iron (Schwartz, 1996).
Pathology
In the full-term newborn, iron stores present at birth are sufficient to prevent iron deficiency during the first 6 months of life. In premature infants, however, iron deficiency can develop in the first few months of life because of low iron
stores at birth. In some infants, intolerance to whole cow’s milk protein can cause a protein losing enteropathy, with occult blood loss leading to severe IDA and hypoproteinemia.
stores at birth. In some infants, intolerance to whole cow’s milk protein can cause a protein losing enteropathy, with occult blood loss leading to severe IDA and hypoproteinemia.
• Clinical Pearl
Infants who consume large amounts of cow’s milk are particularly susceptible to develop IDA because of poor intake of iron-rich solids and a microscopic loss of heme in the gut.
In adolescents, increased requirements during pubertal growth spurt and menstrual blood loss in females are contributing factors to IDA. Less common causes of IDA related to occult blood loss in children include inflammatory bowel disease, peptic ulcer disease, Meckel’s diverticulum, polyps, and hemangiomas. Hookworm infestation and trichuriasis are common causes of IDA in developing countries.
Epidemiology
Iron deficiency remains the most common cause of anemia in children around the world. In the United States, the incidence of IDA in children has decreased considerably due to iron fortification of most infant formulas and the American Academy of Pediatrics’ (AAP) recommendation that bottle-fed infants remain on formula till age 1 year. The incidence of IDA in 12-month-old American infants fed iron-fortified formulas is 0%, compared to 4% in those who received whole cow’s milk and iron-fortified cereals (Fuchs, 1993). In the United Kingdom, the incidence of anemia in 12-month-old infants fed whole cow’s milk from age 6 months was noted to be 31%, compared to only 3% in those who were fed fortified formulas from age 6 months (Daly, MacDonald, Aukett, 1996).
History and Physical Exam
Symptoms of IDA include irritability, pallor, and anorexia. These symptoms usually do not appear until the hemoglobin level is less than 7 gms/dL. Irritability may improve within a few days of starting iron, even before any hematological improvement. In mild cases the physical examination is unrevealing. In more severe cases, the clinician may note pallor, tachycardia, and a systolic flow murmur.
IDA may impair attention span, alertness, and learning in infants and adolescents. These symptoms may not be completely reversible with treatment—hence the importance of prevention.
Diagnostic Studies
The diagnosis of IDA should be strongly considered according to red cell indices and blood smear findings. Studies of plasma iron and storage iron or response to iron therapy can confirm diagnosis. Diagnostic criteria for IDA are listed in Display 42-1.
DISPLAY 42–1 • Iron Deficiency Anemia—Diagnostic Criteria
Suggestive
MCV—decreased proportionate to the degree of anemia. Increased RDW indicates anisocytosis.
Blood smear—microcytosis, hypochromia, anisocytosis, poikilocytosis
Diagnostic
Transferrin saturation—< 16% in children; < 10% in infants. (Serum iron is usually < 40 ug/dL and TIBC > 400 ug/dL.)
Serum ferritin—decreased (< 10 ng/mL)
Response to iron therapy—increase in hemoglobin of at least 1 gm/dL in 2 weeks, and to normal values in 4 to 6 weeks
Management
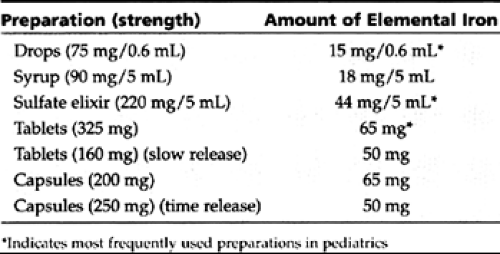
Oral iron in the form of ferrous sulfate or gluconate (3–6 mg/kg of elemental iron per day, in two to three divided doses) is effective in most patients. Providers should become familiar with the contents of a few preparations, all of which are listed in Table 42-9. A reticulocyte response is usually seen within 4 days of beginning therapy. The hemoglobin level begins to rise within a week, reaching normal levels in 4 to 6 weeks.
 |
• Clinical Pearl
After initiating empiric treatment, clinicians must arrange for follow-up. In IDA, a CBC obtained a month after therapy begins will demonstrate a significant rise in hemoglobin or hematocrit, thereby confirming the diagnosis. Iron therapy should continue for an additional 2 months after the patient achieves normal hemoglobin in order to replenish depleted iron stores.
Oral iron therapy is effective in almost all patients. Failure to respond to iron therapy usually indicates ongoing blood loss or poor compliance. Poor tolerance to oral iron or defective gastrointestinal absorption of iron, requiring parenteral iron therapy, is extremely rare in children. Patients should follow the instructions given in Display 42-2.
DISPLAY 42–2 • Patient Instructions for Iron Therapy
Administer iron between meals.
Liquid preparations may stain teeth; rinse with water.
GI symptoms (eg, constipation) are rare.
Antacids and H2 blockers interfere with iron absorption.
In deficiency states, take iron for three months to replenish stores.
Prevention of IDA in infants is of utmost importance. Primary care providers should educate parents regarding the various preventive measures listed in Display 42-3.
DISPLAY 42–3 • Prevention of Iron Deficiency in Infants
Breast-fed infants should receive iron supplements of 1 mg/kg/day from age 6 months.
Non-breast fed infants should receive iron-fortified formula (12 mg of elemental iron per liter) until age 1 year.
Iron-fortified cereals should be started at age 6 months in all infants.
Avoid whole cow’s milk in the first year of life.
Start iron supplements at age 4 weeks in premature babies (2 mg/kg/day).
• Clinical Pearl
Breast milk alone is an insufficient source of iron after age 6 months. Infants exclusively breast-fed should receive iron supplements of 1 mg/kg per day.
Anemia of Lead Poisoning
Lead interferes with heme synthesis, which is essential for erythropoiesis and for basic cellular respiration through the mitochondrial cytochrome system. Thus, lead poisoning can cause microcytic hypochromic anemia in addition to its detrimental effects on the central nervous system. The hematologic effects of lead poisoning, however, do not appear until the lead level is greater than 50 mcg/dL. The microcytic anemia seen in patients with mild lead poisoning is frequently due to associated IDA.
Thalassemias
In thalassemia, partial or complete failure to synthesize globin chains (β or α) results in decreased hemoglobin production. The resulting anemia, therefore, is microcytic and hypochromic. Most individuals with the heterozygous defect have mild microcytic anemia; some have microcytosis with no anemia. In homozygous thalassemia, the synthesis of globin chains is severely impaired, causing imbalance in polypeptide chain production. The polypeptide chains produced in excess precipitate in the cell membrane, causing hemolysis. A high frequency of thalassemia genes is seen in Mediterranean countries, Africa, the Middle East, the Indian subcontinent, and Southeast Asia. From 3% to 8% of Americans of Italian or Greek ancestry and 0.5% of African Americans carry a gene for β-thalassemia (Honig, 1996).
Heterozygous-β thalassemia (β-thalassemia trait)
In this condition, there is marked microcytosis, hypochromia, and poikilocytosis; ovalocytosis and basophilic stippling also may be seen. RDW is normal to slightly elevated. Hb A2 is elevated (>4.0 %) in most patients, and Hb F is slightly increased (2%–6%) in about 50% of cases. In the most common form, β+-thalassemia trait, the anemia is mild, with hemoglobin levels in the range of 9 to 12 gms/dL. Affected children are asymptomatic. In the less common form, β0-thalassemia trait, the anemia may be moderate in severity. Not all patients with the thalassemia trait have microcytosis. Patients with the rare δβ-thalassemia trait have normal MCV, normal or slightly decreased Hb A2 levels, and increased Hb F levels (5%–15%).
Homozygous β0-thalassemia (Cooley’s anemia, Thalassemia major)
Thalassemia major, a severe, progressive, hemolytic anemia, usually presents between ages 6 and 12 months. Clinical features include jaundice secondary to hemolysis and hepatosplenomegaly due to extramedullary hematopoiesis.
The hemoglobin level continues to drop to less than 5 gms/dL. Growth is impaired because of severe anemia, and bone marrow hyperplasia causes head bossing and prominent malar eminences. Unless regular transfusions are given, the life expectancy is no more than a few years. The anemia is microcytic and hypochromic. The blood smear shows many poikilocytes, target cells, and nucleated red cells. Indirect bilirubin and LDH are elevated. Hemoglobin electrophoresis reveals very high levels of fetal hemoglobin, elevated hemoglobin A2, and markedly decreased or no hemoglobin A.
The hemoglobin level continues to drop to less than 5 gms/dL. Growth is impaired because of severe anemia, and bone marrow hyperplasia causes head bossing and prominent malar eminences. Unless regular transfusions are given, the life expectancy is no more than a few years. The anemia is microcytic and hypochromic. The blood smear shows many poikilocytes, target cells, and nucleated red cells. Indirect bilirubin and LDH are elevated. Hemoglobin electrophoresis reveals very high levels of fetal hemoglobin, elevated hemoglobin A2, and markedly decreased or no hemoglobin A.
Monthly transfusions to maintain a hemoglobin level higher than 10gms/dl allow normal growth and development. Within a few years of transfusion therapy, however, chelation (with daily subcutaneous infusion of deferoxamine) is needed to prevent transfusion-induced hemosiderosis. After several years, splenectomy becomes necessary for most patients to decrease the increasing transfusion requirement caused by hypersplenism. Bone marrow transplantation from an HLA-identical sibling can cure most patients. This procedure, however, is not routinely performed because of the risk of morbidity and mortality from bone marrow transplantation.
α-Thalassemia
Four genes—two gene loci on each of chromosome 16—control the synthesis of α chains. Thus, α-thalassemia presents in four degrees of severity according to the number of gene deletions: silent carrier, α-thalassemia trait, Hemoglobin-H disease, and hydrops fetalis (Table 42-10).
 |
α-thalassemia trait usually presents as a mild microcytic anemia that does not respond to iron therapy. Newborns with α-thalassemia trait have elevated levels of hemoglobin Barts (γ4). Southeast Asians commonly carry α0 thalassemia gene mutations, and α+ thalassemia mutations are generally seen in people of African ancestry. Thus, the severe forms of the disease (Hemoglobin-H disease and hydrops fetalis) usually are not seen in African Americans. Nearly 30% of African Americans are silent carriers, and 2% have α-thalassemia trait (Dozy, 1979).
NORMOCYTIC ANEMIAS
Anemia of Inflammation
Acute or chronic inflammation is one of the most common causes of anemia in children. During acute infections, hemoglobin may drop by as much as 2.4 gms/dl, depending upon the severity of the illness.
• Clinical Pearl
Anemia of inflammation may be normocytic or microcytic. Serum iron is low in both anemia of inflammation and iron deficiency.
With chronic inflammation, the release of stored iron from the reticuloendothelial system to the serum decreases; iron stores are normal as evidenced by a normal or high serum ferritin and a low total iron binding capacity (TIBC). Conversely, in IDA, the TIBC is increased, while serum iron and serum ferritin are decreased. The resolution of anemia coincides with that of the inflammation, and may take longer than a month.
• Clinical Pearl
In children, delaying investigation of a mild normocytic anemia by at least 4 weeks is wise when the child has a history of a recent infection. Anemia of inflammation may take this duration to resolve.
Physiologic Anemia of Infancy
In the normal full-term infant, the hemoglobin level drops from a mean of 18.5 gms/dL (-2SD = 14.5 gms/dL) on the first day of life to 11.5 gms/dL (-2SD = 9.0 gms/dL) between ages 8 to 12 weeks. Three factors contribute to this physiologic drop in the hemoglobin, often referred to as physiologic anemia of infancy:
Decreased production of red cells during the first 8 to 12 weeks of life
Decreased life span of fetal red cells
Hemodilution caused by increasing plasma volume associated with rapid growth
Thus it is not uncommon to see normal infants with a hemoglobin of 9 to 10 gm/dl at age 2 to 3 months, especially in association with minor infections. The MCV and RDW are normal. The reticulocyte count may be normal or slightly increased during the recovery phase.
In premature infants, the physiologic anemia occurs earlier, and the degree of anemia is more severe. In infants with birth weight less than 1500 gms, the hemoglobin level drops to 7 to 8 gms at age 3 to 6 weeks. Nutritional deficiencies may exacerbate the physiologic anemia.
Premature infants should receive folic acid (1 mg/day) and iron supplements (2 mg/kg of elemental iron/day) beginning at age 4 weeks.
Transient Erythroblastopenia of Childhood (TEC)
A transient but marked suppression of erythropoiesis, usually following viral infections, causes TEC. The mechanism may be immune-mediated suppression of the erythroid progenitor cells. Parvovirus has been shown to be the etiology in some patients.
The peak incidence is between ages 1 to 3 years. Children with TEC have few symptoms other than pallor at the time of presentation, and physical examination does not reveal any abnormal findings except for pallor and tachycardia. The anemia is usually severe, with hemoglobin levels less than 8 gm/dl. The red cell count is decreased; MCV, MCH, and RDW are normal. The reticulocyte count is less than 1% in most patients. In some patients, the absolute neutrophil count (ANC) may be less than 1000/mm3 at diagnosis (Cherrick, 1994). The platelet count is increased in most patients, especially during the recovery phase. Most children recover within one to two months from diagnosis. About 5% to 10% of patients are in the recovery phase when they first see the provider. These patients have normal or increased reticulocyte counts and elevated MCV.
Diagnosis of TEC is based on excluding other causes of anemia; spontaneous recovery confirms the clinical impression. Diamond-Blackfan syndrome (congenital pure red cell aplasia) should be suspected in the presence of the following features: age younger than 6 months, macrocytosis, increased HbF, and lack of recovery within 2 months. No specific therapy is available for TEC. Transfusion of red cells is indicated only if there is evidence of cardiac decompensation secondary to the severe anemia. Recurrence of TEC is rare.
MACROCYTIC ANEMIAS
Megaloblastic Anemias
Both vitamins B12 and folate are essential for DNA synthesis, and a deficiency of either or both will cause failure of DNA synthesis and disordered cell proliferation. The increased interval between cell divisions allows more cell growth but fewer divisions, leading to macrocytosis. In the bone marrow, the macrocytosis is most apparent in the erythroid precursors. The megaloblastic picture is characterized by a nucleus that looks less mature than the cytoplasm; the nuclear chromatin appears stippled and lacy, while the cytoplasm appears normally mature and hemoglobinized.
Folic Acid Deficiency
Folate is abundant in many foods including whole wheat, green vegetables, carrots, fruits, egg yolk, and animal organs (eg, kidney, liver). The normal adult requirement of folate is about 100 mcg /day. The causes of folate deficiency in children are given in Display 42-4.
DISPLAY 42–4 • Causes of Folate Deficiency
Low birth weight
Deficient intake: goat’s milk, powdered milk
Defective absorption:
Malabsorption syndromes: Celiac disease, kwashiorkor, marasmus, chronic infectious enteritis, enteroenteric fistulas
Congenital folate malabsorption
Increased requirements: pregnancy, hemolytic anemia
Drugs:
Anticonvulsant drugs: phenytoin, primidone, phenobarbital
Methotrexate, trimethoprim, oral contraceptives pyrimethamine
Congenital dihydrofolate reductase deficiency
Human milk and cow’s milk provide adequate amounts of folic acid; goat’s milk and powdered milk are poor sources of folate. Babies receiving only goat’s milk require supplementation, which usually can be achieved with a multivitamin containing the B group.
• Clinical Pearl
Serum folate levels drop within 2 weeks after cessation of folate intake. Red cell folate, which better represents body folate stores, may remain within normal limits for up to 3 months of deficient folate intake.
History and Physical Exam
The peak incidence for megaloblastic anemia of infancy is ages 4 to 7 months. Irritability, poor weight gain, and chronic diarrhea are common symptoms. Infants with congenital folate malabsorption present with mouth ulcers, progressive neurologic deterioration, and immune deficiency in addition to the above symptoms. In infants with poor nutrition, IDA may coexist.
Diagnostic Criteria and Studies
MCV is markedly increased (>100). The blood smear shows hypersegmented neutrophils and marked anisocytosis and poikilocytosis of red cells. The reticulocyte count is decreased. Neutropenia and thrombocytopenia may be present in some cases. Serum LDH is markedly elevated. Bone marrow shows erythroid hyperplasia with megaloblastic changes and giant metamyelocytes.
The diagnosis is confirmed by a serum folate less than 3 ng/mL (normal range 4–20 ng/mL) and a red cell folate less than 150 ng/mL (normal range 200–800 ng/mL). The provider should exclude concomitant presence of B12 deficiency before starting treatment.
Management
Folic acid in a dose of 100 mcg/kg per day, given orally or parenterally for 3 to 4 weeks, is sufficient for most patients. Larger doses of folic acid can exacerbate neurologic damage in patients with cobalamin (B12) deficiency. Patients with congenital folate malabsorption will require intramuscular folinic acid (5-formyl tetrahydrofolate) therapy, which should be instituted early to prevent neurologic deterioration.
Vitamin B12 Deficiency
Vitamin B12 is a cobalt containing porphyrin, named cobalamin. It is found in foods of animal origin (eg, liver), fish, and dairy products, but not in fruits, cereals, or vegetables. A normal diet contains a large excess of B12 in comparison to daily needs. B12 combines with intrinsic factor (a glycoprotein
synthesized by the gastric mucosa) and is absorbed to the receptors in the distal ileum. B12 alone is then absorbed into the portal blood, where it attaches to transcobalamin II and is transported to the bone marrow and other tissues. The total body content of vitamin B12 is 3 to 5 mg, and the rate of daily loss of vitamin B12 is less than 0.1% of the total body stores.
synthesized by the gastric mucosa) and is absorbed to the receptors in the distal ileum. B12 alone is then absorbed into the portal blood, where it attaches to transcobalamin II and is transported to the bone marrow and other tissues. The total body content of vitamin B12 is 3 to 5 mg, and the rate of daily loss of vitamin B12 is less than 0.1% of the total body stores.
• Clinical Pearl
As opposed to nutritional folate deficiency, which becomes clinically apparent within months, it takes more than 2 years of poor absorption of vitamin B12 before clinically significant B12 deficiency develops (Hughe-Jones & Wickramasinghe, 1991).
Deficiency of vitamin B12 occurs in various clinical conditions (see Display 42-5). Juvenile pernicious anemia is a megaloblastic anemia caused by absence of effective intrinsic factor (IF). In some patients, IF is present but functionally defective. Gastric acid secretion is normal.
DISPLAY 42–5 • Causes of Vitamin B12 Deficiency
Intrinsic factor (IF) deficiency (juvenile pernicious anemia)
Transcobalamin II deficiency
Intestinal malabsorption of B12
Familial absence of receptor for IF-B12 (Imerslund-Grasbeck syndrome)
Surgical resection of terminal ileum
Crohn’s disease
Diverticulitis
Diphyllobothrium latum (fish tape worm) infestation
History and Physical Exam
The presenting symptoms may be those of severe anemia (ie, weakness, listlessness, anorexia, and irritability) or of neurologic involvement (ie, failure to reach milestones, slow mental development, parasthesias, hyporeflexia, ataxia, extensor plantar reflexes, and clonus). Most children with juvenile pernicious anemia present between 1 to 5 years of age.
Diagnostic Criteria and Studies
Peripheral blood picture and bone marrow findings are identical to that seen in folate deficiency (see above). Serum B12 levels are usually decreased (<100 pg/mL) but by themselves are not diagnostic. A Schilling test differentiates juvenile pernicious anemia (IF deficiency) from other causes of vitamin B12 deficiency.
Plasma levels of methylmalonic acid and total homocysteine are markedly increased. Providers should routinely obtain these levels when they suspect B12 deficiency, since serum B12 levels can be normal in the presence of significant B12 deficiency.
Management
Initial treatment consists of an intramuscular injection of 1 mg of vitamin B12. Children with neurologic involvement should receive 1 mg of B12 daily for 2 weeks. They will need maintenance doses of 1 mg given monthly for life.
Aplastic Anemia (Pancytopenia)
Acquired Aplastic Anemia
Pancytopenia resulting from bone marrow failure is referred to as aplastic anemia. Acquired aplastic anemia may result from direct damage to the stem cell or bone marrow micro-environment or from immune-mediated insult to the progenitor cells. Known causes include drugs, toxins, and viruses (Display 42-6). Patients present with symptoms of severe anemia, bleeding due to thrombocytopenia, or frequent infections secondary to neutropenia. The aplastic anemia is considered severe when two of the following three criteria are present: absolute neutrophil count (ANC) <500/mm3, platelet count <20,000/mm3, or reticulocyte count <1% after correction for hematocrit.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


