Key Clinical Questions
What are the “classic” clinical and basic laboratory features of each of the common hematologic malignancies? (“How do I prove the diagnosis?”)
What is the natural history of each disease entity without therapy?
What is the goal of treatment for each disease entity (cure, prolongation of survival, or purely palliation)?
Which modalities may be employed for each disease entity (chemotherapy, immunotherapy, radiation, stem cell transplant)?
What is the prognosis with therapy? How can the prognosis estimate be refined?
What needs to be done to prepare the patient for therapy (to communicate risks and benefits and to reduce toxicity)?
How do we assess the response?
How do we monitor the patient after completion of therapy, both early on (for relapse) and later (for delayed toxicities of therapy)?
Section 1: The Concept of Malignancy
Practitioners need to be familiar with the broad classes of hematologic malignancy (acute and chronic leukemias, aggressive and indolent lymphomas, myeloma, myeloproliferative neoplasms, and myelodysplastic syndromes). They should also be familiar with the side effects of therapy, for example, acute and chronic cardiotoxicity from anthracyclines, long-lasting immunosuppression from purine analogs, and the expected timing of neutropenia (Table 179-1). He or she should also be comfortable managing a number of hematologic emergencies, which are described in the following chapter. The specifics of diagnosis and treatment can be both subtle and complex. Subspecialty consultation with a hematologist or oncologist is essential in caring for a patient who is suspected or known to suffer these conditions.
| Drug | Mechanism | Uses | Side Effects | Comments |
|---|---|---|---|---|
| Chemotherapy and targeted therapy | ||||
| All-trans retinoic acid (ATRA) and arsenic trioxide (ATO) | Differentiating agents | Acute promyelocytic leukemia |
| Dexamethasone 10 mg IV twice a day should be started at the earliest sign of differentiation syndrome, even if other etiologies like sepsis or primary CHF are possible. In patients with acute renal failure or requiring ICU admission for respiratory distress, ATRA or ATO should be temporarily held. |
| Anagrelide | A prostaglandin synthetase inhibitor, specifically inhibits platelet production but occasionally causes mild anemia | Myeloproliferative neoplasms | Palpitations, CHF, noncardiac edema, diarrhea, abdominal pain, headache, bleeding, anemia | In the MRC PT-1 trial, aspirin + hydroxyurea were compared to aspirin + anagrelide treatment for ET. There were more arterial events and fewer venous thromboses in the anagrelide arm. Anagrelide increased progression to marrow fibrosis and conferred a higher risk of serious hemorrhage. More patients withdrew from the anagrelide arm due to adverse effects. A starting dose of 0.5 mg twice a day is appropriate. |
| Anthracyclines (daunorubicin, doxorubicin, idarubicin, epirubicin) |
| Acute leukemias (eg, daunorubicin in “3 + 7”), lymphomas (eg, doxorubicin is the H in “CHOP”). | Myelosuppression. Early cardiotoxicity may manifest as transient asymptomatic sinus tachycardia or ECG abnormalities and does not predict late cardiotoxicty. Late cardiotoxicity is dose-dependent. Extravasation may cause tissue necrosis days to weeks later. Radiation recall, high emetogenicity, reversible alopecia, and secondary leukemia are other considerations. | Baseline assessment of ejection fraction is advised. Anthracyclines are contraindicated if the EF is < 45%, if the patient suffered an MI in the past three months, or in the presence of severe arrhythmias. Cumulative lifetime doses are limited to 450–550 mg/m2 doxorubicin, 800 mg/m2 daunorubicin, 150 mg/m2 idarubicin. |
| 5-Azacitidine and decitabine | At low doses, they act as demethylating agents (inhibit DNA methyltransferase) | Myelodysplastic syndromes | Both drugs cause nausea, diarrhea, constipation, and cytopenias. SC injection site reactions, neuropathy, hepatotoxicity, rash, hypokalemia, and renal failure have occurred with 5-aza. | Profound cytopenias may herald an eventual response to treatment rather than progressive disease. |
| Bleomycin | A glycopeptide antibiotic, it causes scission of single-and double-stranded DNA and interferes with DNA repair | Hodgkin lymphoma, uncommonly used today in NHL | Fever and chills occur in half of patients within hours of treatment. Anaphylactoid reactions may occur. Interstitial pneumonitis and pulmonary fibrosis may be life threatening. The presentation may be subtle: nonproductive cough, dyspnea, unexplained fever. CXR usually shows bilateral reticular or nodual opacities without pleural effusions, but may be normal, and reduced DLCO is the first PFT abnormality. Flagellate erythema is a rare but distinctive rash that may appear from one day to several months following bleomycin administration. Bleomycin enhances radiation injury. Alopecia. | Bleomycin should be withheld if the DLCO falls to 30–35% of the initial value, if the FVC falls significantly, or if there are clinical or radiographic features suggesting pulmonary toxicity. Prednisone 1 mg/kg should be employed in patients with pneumonitis. Since oxygen may synergize with bleomycin to produce fibrosis, supplemental oxygen administered during operative procedures and critical illnesses should be minimized. |
| Bortezomib | A proteosome inhibitor | Multiple myeloma. Investigational and salvage use in certain lymphomas. | Peripheral neuropathy that is sometimes painful, orthostatic hypotension, gastrointestinal upset, constipation, ileus, thrombocytopenia, herpes zoster, tumor lysis syndrome. | Zoster prophylaxis is advisable in VZV+ patients. The neuropathy may require dose reductions and usually improves or resolves within two years of discontinuing treatment. |
| Busulfan | An alkylating agent, it forms DNA intrastrand and interstrand crosslinks. | Conditioning for allogeneic stem cell transplant. At lower doses, it may be used for palliation in myeloproliferative neoplasms and CML. | Myelosuppression may be prolonged. An Addisonian-like syndrome with skin hyperpigmentation and weakness but normal adrenal function may occur. Alveolar and interstitial pneumonitis are well described. Seizures may occur in the setting of transplant conditioning and busulfan may potentiate cyclophosphamide in causing sinusoidal obstruction syndrome. | In the transplant conditioning setting, drug level monitoring with dose adjustment to achieve 800–1000 ng/mL reduces serious toxicity and phenytoin or, less commonly, lorazepam seizure prophylaxis is employed. Pulmonary toxicity is treated with steroids (eg, prednisone 50–100 mg/day) and minimizing inspired oxygen. |
| Chorambucil | An alkylating agent | CLL, low-grade lymphoma | Generally well-tolerated. Myelosuppression is the usual dose-limiting toxicity. Pulmonary toxicity, rash, and toxic epidermal necrolysis are rare occurrences. | CBCs should be monitored closely, especially with continuous administration. |
| Cyclophosphamide | An alkylating agent, it interferes with DNA replication and transcription of RNA | Lymphoma, transplant conditioning | Myelosuppresion is the major dose-limiting toxicity. Hemorrhagic cystitis is common (10%, up to 40% with high doses). Tumor lysis syndrome may occur. Nasal stuffiness and facial discomfort are common complaints with rapid infusions. Pulmonary toxicity occasionally occurs and has a high fatality rate. SIADH is seen. In the transplant setting, Cy is the main drug responsible for sinusoidal obstruction syndrome and cardiac necrosis can occur at high doses. Alopecia, radiation recall, and secondary leukemia and other cancers are other considerations. | Patients should be well hydrated and prophylactic mesna is used to minimize the occurrence of hemorrhagic cystitis. When this complication does supervene, treatment includes aggressive hydration (to ensure a diuresis of 3 L/24 h) and maintaining platelets > 50 × 109/L. If the urine does not clear, pain is severe, or renal insufficiency develops, then obtain a renal ultrasound and start continuous bladder irrigation adding 300 mg of hydrocortisone to each 3 L bag of irrigation fluid. Additional measures include cystoscopic evacuation of clots and fulguration, and intravesical carboprost. |
| Cytarabine | An analogue of cytidine, AraC inhibits DNA polymerase, thus stopping DNA elongation. It is also incorporated into DNA leading to errors in replication and transcription. | AML induction and consolidation, MDS, high-grade lymphomas | Myelosuppression. Cytarabine sometimes provokes a generalized rash, fever, and myalgia 6–12 hours after administration of high doses. Palmar–plantar erythrodysesthesia manifests as painful paresthesias and edematous erythema of the palms and soles, followed weeks later by desquamation. Rash is due to build up of the drug in areas that lack sweat glands, not an allergic reaction, and further treatment with this agent is permissible. Chemical conjunctivitis is common with high doses and prophylactic steroid eye drops are required. Cerebellar degeneration can occur and may be irreversible. Hepatotoxicity and pulmonary toxicity may occur. | Test for cerebellar dysfunction (include handwriting) before each high dose infusion used in consolidation therapy. Cerebellar toxicity mandates cessation of this therapy. Steroids may be used to treat pulmonary toxicity, which may present as a capillary leak syndrome 2–21 days after the first dose. |
| Fludarabine | A purine analogue, it inhibits DNA and RNA polymerase, DNA primase, DNA ligase and ribonucleotide reductase | CLL, lymphomas, salvage regimens for AML, conditioning regimens in nonmyeloablative stem cell transplantation | Myelosuppression may persist for many months. Profoundly reduces B and T lymphocytes; this degree of immunosuppression can lead to opportunistic infection with intracellular pathogens (PCP, Listeria) and transfusion-related graft-versus-host disease. Interstitial pneumonitis and dose-dependent irreversible neurotoxicity (including confusion, cortical blindness, and necrotizing leukoencephalopathy) rarely occur in current protocols. Autoimmune haemolytic anemia is common when used in DAT+ CLL patients. | Blood banks and patients themselves should be aware of the need to irradiate cellular blood products. PCP prophylaxis is required. |
| Hydroxyurea | Considered an antimetabolite, it inhibits ribonucleotide reductase, which leads to depletion of the deoxynucleoside triphosphate pool. | Myeloproliferative neoplasms, CML, temporary control of AML | Dose-related neutropenia, macrocytic anemia, thrombocytopenia. Vasculitic ulcerations in patients with MPNs. Mouth ulcers. Enhances the toxicity of radiation and may cause radiation recall. | Hydroxyurea will ameliorate splenomegaly and hypercatabolic symptoms. Women on chronic hydroxyurea therapy who are contemplating pregnancy should be switched to interferon-alpha. |
| Imatinib (first generation), nilotinib, dasatinib (second generation) | Tyrosine kinase inhibitors with varying specificity for BCR-ABL and other kinases | CML, Ph+ ALL, and subsets of CMML, chronic eosinophilic leukemia, and systemic mastocytosis |
| The three currently available TKIs have very different drug-drug interactions and side effect profiles. Toxicity from one agent does not preclude switching to another TKI. |
| Glucocorticoids (eg, prednisone, dexamethasone, methylprednisolone) | Upregulate or downregulate gene transcription, and cause apoptosis of lymphocytes. | ALL, CLL, NHL, Hodgkin lymphoma, myeloma | Short-term: Euphoria, psychosis, insomnia, hyperglycemia, appetite stimulation, myopathy, fluid retention, hypertension, hypokalemia. Chronic use: Cushingoid appearance, peptic ulcers, osteoporosis, avascular necrosis, cataracts, impaired cellular immunity. | Side effects should be actively managed. Fungal and PCP prophylaxis may be indicated, depending on dose, duration of use, and prior history of candidal or mold infections. In patients in whom a prolonged (≥ 3 month) course of glucocorticoids is envisioned, primary osteoporosis prevention with a bisphosphonate in addition to calcium and vitamin D should be considered, especially if the baseline T-score is ≤ -1. |
| L-asparaginase | By hydrolyzing asparagine, it deprives lymphoid cells of this required amino acid, impairing protein synthesis. | ALL | Hypersensitivity reactions, acute pancreatitis, hyperglycemia, hepatotoxicity. Thrombosis is frequent but hemorrhage may also occur, as asparagine depletion interferes with liver synthesis of both anticoagulant and procoagulant proteins. Lethargy, confusion, and personality changes resolve within days of drug discontinuation. | For allergic patients, Erwinia-derived asparaginase should be substituted for standard E Coli-derived asparaginase. Asparaginase is poorly tolerated in adults, particularly because of thrombotic complications. Serum amylase should be obtained prior to each cycle, and significant elevations or symptomatic pancreatitis preclude further therapy with this drug. |
| Lenalidomide and thalidomide | Immunomodulators with antiangiogenic activity | Myeloma (both). 5q- syndrome/MDS (lenalidomide). Investigational use in many other hematologic cancers. |
| The risk of thrombosis is augmented by concurrent glucocorticoids or erythropoietin. The optimal VTE prophylaxis is debated. |
| Melphalan | An alkylating agent | Low dose: elderly or unfit patients with myeloma; High-dose: conditioning for autologous stem cell transplant in myeloma, NHL, and HL | Mucositis is the limiting effect of high dose treatment. Pneumonitis and pulmonary fibrosis rarely occur. Tissue necrosis occurs with extravasation. | Prolonged alkylator exposure precludes subsequent autologous stem cell collection, which is why melphalan is avoided as initial therapy in transplant-eligible patients. |
| 6-Mercaptopurine | A purine analogue | Maintenance therapy in ALL and APL | Myelosuppression, especially in patients with thiopurine methyltransferase deficiency; hepatotoxicity, immunosuppression, radiation recall. | Allopurinol significantly increases plasma concentrations of 6-MP. When administered together, the 6-MP dose should be reduced by 25–33%. 6-MP should be stopped promptly at the first indication of myelosuppression, since the nadir may be delayed. |
| Methotrexate | As an antifolate, it prevents de novo purine and pyrimidine biosynthesis. | High-grade NHL, ALL, prevention of graft-versus-host disease in the transplant setting | Myelosuppression, stomatitis, mucositis, tumor lysis syndrome, hepatotoxicity, acute renal insufficiency, idiosyncratic pulmonary toxicity, radiation recall, chemical meningitis with intrathecal administration. | Many drugs can reduce renal clearance of MTX or displace MTX from albumin, increasing bioavailability. For example, penicillins should not be administered with high-dose MTX. MTX also accumulates in effusions and edema fluid, which can lead to substantial toxicity. Effusions should be evacuated before MTX is given. Leucovorin rescue, hydration, and urinary alkalinization are the mainstays of treating MTX toxicity. High flux hemodialysis may be tried, and glucaripidase may be available on a compassionate basis. |
| Vinca alkaloids (vincristine and vinblastine) | Antimicrotubule agents | ALL, NHL, Hodgkin lymphoma, myeloma | Vincristine’s dose-limiting effect is neurotoxicity. Commonly, this includes peripheral sensory impairment, paresthesias, and loss of deep tendon reflexes. Painful dysesthesia, motor impairment that includes foot drop and difficulty with fine finger movements, ataxia, cranial nerve palsies manifesting as hoarseness, ptosis, facial nerve palsies, or jaw pain, and in extreme cases paralysis can occur. Autonomic toxicity includes constipation and ileus. Vinblastine is less neurotoxic. Both drugs are vesicants. | With vincristine, aggressive bowel regimens are needed to prevent obstipation and ileus. Extravasation may be treated by infiltrating the area with 1–2 mL of hyaluronidase, 150 U/mL, then applying warm compresses for 72 hours. |
| Immunotherapy | ||||
| Alemtuzumab | A monoclonal anti-CD52 antibody | CLL, T-cell lymphomas | Infusion reactions, as described for rituximab below, are common during the first week of therapy. Profound cytopenias are very common. Opportunistic infections due to profound lymphopenia are frequent. | Prophylaxis against PCP and herpetic infections, and prophylactic or preemptive therapy for CMV is mandatory. Blood products administered prior to recovery from lymphopenia should be irradiated to avoid transfusion-related graft-versus-host disease. |
| Rituximab | A monoclonal anti-CD20 antibody | CLL, B-cell NHL | Infusion reactions including fevers, rigors, urticaria, bronchospasm and hypotension are common. The incidence of infusion reactions is highest with the first dose (77%) and diminishes with subsequent doses. Anaphylaxis may occur, usually at the second or subsequent infusion. Pulmonary reactions that manifest with dyspnea, bronchospasm, hypoxia, and lung infiltrates may occur acutely (one to two hours after initiation of the first infusion) or be delayed (one to four weeks after infusion). Tumor lysis syndrome is well-recognized (particularly in CLL). Due to immunosuppression, hepatitis reactivation and extremely rarely progressive multifocal leukoencephalopathy may occur; the risk of PML is likely augmented when rituximab is combined with chemotherapy. | Most infusion reactions are mild-to-moderate and can be managed by interrupting or slowing the infusion and supportive care (saline, antihistamines, methylprednisolone, acetaminophen, bronchodilators, ± epinephrine if severe). |
In general, each nucleated somatic cell belonging to a given person contains an identical DNA sequence. The exception occurs in normal lymphocyte development, in which the actual DNA sequences encoding immunoglobulin genes (in the case of B cells) or the T cell receptor (in the case of T cells) are altered. Because different cell types do not derive their unique features from differences in the DNA sequence, the regulation of gene expression is crucial to ensure that growth, proliferation, cell-specific functions, and cell death occur at the appropriate times in the life of the cell.
Clonal proliferation is the sine qua non of malignancy. Clonal proliferation refers to the concept that an entire population of malignant cells arises from a single ancestral cell that, due to one or more genetic or epigenetic changes, develops the ability to proliferate autonomously, independent of physiologic growth stimuli. The cancer-initiating cell is often conceptualized as a “cancer stem cell,” but whether the inciting genetic mutations actually occur in a normal stem cell or in a more mature cell that then reactivates primitive or “embryonic” gene expression programs, and so regresses in differentiation, is a matter of active investigation. The hallmark of a stem cell is its ability to “self-renew” an unlimited number of times. Self-renewal may be accomplished through symmetric cell division in which the stem cell gives rise to two identical daughter stem cells, or through asymmetric cell division in which the stem cell divides in a way that yields one copy of itself and one daughter cell that will launch a differentiation program.
Two basic mechanisms of genetic alteration contribute to neoplastic transformation: mutations in the DNA sequence, and chromosomal aberrations. Genomic instability is an important consequence of DNA mutations and chromosomal aberrations. Fusions may corrupt transcription factors that would otherwise activate genes needed for DNA mismatch repair and for repair of double strand breaks. The p53 protein normally pauses the cell cycle long enough for DNA repair to occur or if the damage is extensive, directs the cell toward apoptotic cell death. In hematologic malignancies, as in solid tumors, p53 function is often sabotaged. This allows accumulation of additional defects in tumor suppressor and oncogenic pathways, which leads to continued evolution of the malignant clone and favors the emergence of more aggressive subclones.
Although clonal proliferation is necessary for malignancy, it is not sufficient. Secondary clonal events due to genomic instability must lead to loss of cell cycle control, increased self-renewal, and escape from apoptosis. Dissemination of malignant cells is facilitated by down-regulation of adhesion molecules that anchor cells to their stromal niches, up-regulation of surface molecules that promote interaction with the endothelium, and expression of particular cytokines to promote extravasation. The neoplastic cells must also evolve mechanisms to escape immune surveillance.
Lymphoid neoplasms Lymphoid neoplasms appear to recapitulate stages of normal B cell or T cell differentiation, so they are largely classified according to the normal counterpart of the neoplastic cell. For example, B cell neoplasms can be related to one of four stages: (1) precursor B cells, (2) pregerminal center B cells, (3) germinal center B cells (centroblasts and centrocytes), and (4) post-germinal center B cells (memory B cells and plasma cells).
Classification of lymphoid neoplasms is not based on any single parameter. Morphology and various surface markers in combination (as detected by immunohistochemistry or flow cytometry) usually suffice to render a diagnosis. The subtleties in refining the diagnosis are complex; morphology can be atypical and no surface marker is 100% sensitive and specific for any particular lymphoid neoplasm. Chromosome translocations, overexpression of particular gene products (like the ALK enzyme or bcl2), rearrangement of the immunoglobulin genes or TCR, and detection of viral components (eg, Epstein-Barr virus, human T cell leukemia/lymphoma virus) are considered when they are appropriate to the pathologic differential diagnosis.
Myeloid neoplasms The acute and chronic myeloid leukemias likewise can be classified according to the normal counterpart that most closely resembles them in morphology and immunophenotype. The analogy is not ideal, as most myeloid leukemias likely arise from a primitive hematopoietic stem cell or progenitor cell, even if the ultimate differentiation block produces a more mature-appearing neoplasm. By contrast, the lymphoid neoplasms might have actually arisen from the presumed normal counterpart. The French-American-British classification first produced in 1976 and updated through 1989 distinguished eight subtypes of acute myeloid leukemia based on morphology and simple immunohistochemical stains. These include M0 (minimally differentiated), M1 (without maturation), M2 (with granulocytic maturation), M3 (promyelocytic), M4 (myelomonocytic), M5a (monoblastic) and M5b (monocytic), M6a (erytholeukemia) and M6b (pure erythroid leukemia, which is extremely rare), and M7 (megakaryocytic). The FAB also identified subtypes of myelodysplastic syndrome (eg, refractory anemia) and myeloproliferative disorders (eg, polycythemia vera). FAB terms are still widely used in common speech, but the current standard classification was issued by the World Health Organization and most recently updated in 2008. The WHO classification of myeloid neoplasms relies upon surface markers, genetic aberrations, clinical presentation and prognosis, and any relation to known inciting factors.
The terms “lymphoma” and “leukemia” are often confusing. In general, “leukemia” means there are cancerous cells of hematopoietic origin in the bone marrow and usually also circulating in the peripheral blood. These cells may be of myeloid or of lymphoid origin. “Lymphoma” means that there are tumors of lymphoid origin causing enlargement of lymph nodes or spleen, or causing masses in organs like the liver or gut.
More precisely, the exact definition of leukemia versus lymphoma depends on the clinical and pathologic features of specific disease entities. For example, in acute lymphoblastic diseases, some treatment protocols use the term “leukemia” if malignant blasts comprise more than 25% of nucleated cells in the bone marrow and the term “lymphoma” if there are fewer than 25% marrow blasts, regardless of whether the patient bears other masses, such as enlarged lymph nodes or tumors in the anterior mediastinum. This 25% cut-off is arbitrary. The current WHO classification advises using the term “lymphoblastic leukemia” when marrow and blood involvement is extensive. In acute myeloid leukemias, the WHO considers 20% marrow involvement to represent the lowest threshold at which to diagnose “leukemia” as opposed to “myelodysplasia” (barring certain genetic aberrations that flip the diagnosis to “leukemia” even at lower proportions of blasts). By contrast, in the WHO classification, there is no “lower threshold” for bone marrow involvement in the diagnosis of acute lymphoblastic leukemia.
As another example, for CLL/SLL, we use the term “chronic lymphocytic leukemia” if there are more than 5 × 109/L circulating malignant B cells with or without lymph node, spleen, or liver enlargement, but we use the term “small lymphocytic lymphoma” if there is lymphadenopathy (or spleen or liver enlargement) with fewer than 5 × 109/L circulating malignant lymphocytes and no cytopenias attributable to marrow infiltration. Finally, if the blood shows fewer than 5 × 109 cells/L malignant lymphocytes and nodal and organ enlargements are absent, we diagnose “monoclonal B lymphocytosis of unknown significance” (MBUS). MBUS can be found in 3–4% of healthy adults over age 40 and occasionally evolves to CLL.
On the other hand, lymphadenopathy and “masses” are not unique to neoplasms of lymphoid origin. Myeloid leukemias do occasionally form masses and do occasionally cause lymph node enlargement. Masses of myeloid blasts infiltrating organs (other than bone marrow) and effacing normal tissue architecture are called “myeloid sarcomas.” (Old terms include “granulocytic sarcoma” and “chloroma.”) Simple enlargement of the spleen due to infiltrating AML or simple infiltration of the skin or gingival without effacement of the normal tissue architecture would not be considered a myeloid sarcoma.
In the course of establishing a diagnosis, hematologists increasingly rely on advanced molecular laboratory methods to detect DNA mutations, chromosomal aberrations, and chimeric proteins. For example, polymerase chain reaction (PCR) can be used to detect jak2 harboring the V617F mutation that characterizes most cases of polycythemia vera. Standard G-banded karyotypes often reveal chromosomal aberrations of diagnostic importance. Fluorescence in situ hybridization (FISH) can be used to detect fusion genes such as BCR-ABL in CML or PDGFRβ-FLIPI in chronic myelomonocytic leukemia. Several techniques are available to study the immunoglobulin genes and the T cell receptor in order to prove clonality of suspected lymphoid malignancies. PCR or immunohistochemistry can also be used to detect certain viruses (like Epstein-Barr virus or human T cell leukemia/lymphoma virus) that are associated with particular lymphoid cancers.
The results of such investigations often influence treatment decisions based upon their prognostic properties. For example, certain chromosomal aberrations, such as t(8;21) and inversion of chromosome 16, confer a good prognosis in AML. Consequently, hematologists would not recommend frontline allogeneic stem cell transplants to these patients. If acute promyelocytic leukemia is associated with t(15;17) (q22;q12) fusing PML to RARα, the patient is likely to respond well to all-trans retinoic acid (ATRA). Other chromosomal aberrations, such as loss of part of chromosome 5 or 7, or inversion of chromosome 3, confer a dismal prognosis in AML. Hematologists would routinely offer allogeneic stem cell transplant to young, fit patients with these karyotypes. DNA sequence mutations can also inform prognosis. For example, a particular type of mutation in the Flt3 tyrosine kinase (internal tandem duplication in the DNA sequence encoding the juxtamembrane domain of the molecule) confers a poor prognosis in AML.
When a clinician performs a bone marrow aspiration in a patient suspected of harboring an undiagnosed hematologic malignancy, three draws should normally be performed: one for histopathology smears (prepared at the bedside or drawn into EDTA for immediate preparation in the lab), one for flow cytometry (sodium heparin or EDTA tube, per institutional guidelines), and one for cytogenetics/molecular testing (sodium heparin tube). For the smears and flow cytometry samples, only 0.5–1 mL should be withdrawn with each pull to avoid diluting the sample with blood. The bone marrow biopsy is useful to assess cellularity, infiltration with carcinoma, and fibrosis. Although not all hospital laboratories can perform the more specialized tests, the samples can normally be sent to reference centers.
Acute myeloid leukemia (AML) occurs in 3.5 persons per 100,000 per year. It comprises 80–90% of adult acute leukemias. Acute lymphoblastic leukemia (ALL) occurs in 1 person per 100,000 per year. It comprises only 10–20% of adult acute leukemias but accounts for the majority of childhood leukemia.
Chronic myeloid leukemia (CML) accounts for 15% of all adult leukemias. Its global incidence is 1.6 per 100,000 persons per year. The median age at diagnosis is 53 years and the incidence increases with age, although CML can even occur in childhood. While ionizing radiation is associated with CML and its incidence is increased among survivors of the Hiroshima nuclear bomb, most cases have no known cause. The incidence of chronic lymphocytic leukemia (CLL) in Western countries is 2–6 per 100,000 persons per year. The incidence rises sharply with increasing age; above 75 years, the incidence is 35 to 40/100,000 persons per year. The median age at diagnosis is 72 years. There is a 2:1 male to female predominance. CLL is not associated with radiation exposure.
The median age at diagnosis is around 70 years. While the crude incidence is around 2 per 100,000 persons annually, the age-adjusted incidence is around 20 per 100,000 persons over age 70. Ten percent to 15% of cases are estimated to be related to prior chemotherapy or radiation.
The incidence of each of the major non-CML myeloproliferative neoplasms (MPNs) ranges from 0.2 to 3 cases per 100,000 persons per year. They can occur at any age but are more common in older adults. Essential thrombocythemia (ET) shows a second peak in younger women in the third to fifth decade, and these women usually suffer few complications. Familial predisposition to adult-onset MPNs is also described. Radiation exposure has been linked to polycythemia vera (PV).
NHL is the fifth most common cancer in adults, with a crude incidence of around 20 per 100,000 persons per year. Its incidence rises with age. For unknown reasons, its incidence in the Western hemisphere and Australia has been rising at a rate of 2–3% per year for the past three decades.
HL is uncommon with an annual incidence of 2.5 per 100,000 persons. The peak incidence is around age 30, but there is a second, smaller peak in adults older than 65 years.
Monoclonal gammopathy, detected as an abnormal spike on serum protein electrophoresis, is common. In the absence of cytopenias (anemia), renal insufficiency, hypercalcemia, lytic lesions attributable to plasma cell expansion, or conditions such as cryoglobulinemia or cold agglutinin disease, the presence of an asymptomatic monoclonal component is termed “monoclonal gammopathy of undetermined significance” (MGUS). MGUS was detected in 3% of persons 50 years of age or older, 5% percent of persons 70 years of age or older, and 7.5% of those above age 85 in a predominantly Caucasian cohort. The prevalence is about three-fold higher among African Americans. MGUS evolves to myeloma, related plasma cell dyscrasias, or less frequently to lymphoma at a rate of approximately 1% per year, but the risk can be more precisely quantified for the individual patient by taking into account the immunoglobulin isotype (IgG vs. non-IgG), the size of the monoclonal peak (risk increasing above 15 g/L), and the serum free light chain ratio. It is thought that most myeloma evolves from antecedent MGUS.
The incidence of myeloma is 5.4 per 100,000 persons per year, but it is twice as high among African Americans, who have higher physiologic immunoglobulin levels and a higher prevalence of MGUS. Like MGUS, the incidence of myeloma increases with age.
Section 2: The Acute Leukemias
This section will discuss acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) together, comparing and contrasting them at times, because the salient clinical features are similar in adults. The acute leukemias are very aggressive hematologic cancers, are usually fatal within weeks if not properly treated, and require urgent inpatient management. The goal of care in young, fit patients is to cure the disease with chemotherapy and possibly stem cell transplantation. In elderly patients (above age 60), outcomes are poor, with less than 10% survival in those with unfavorable cytogenetics, even with the most aggressive therapy. In this population, palliation with periodic “gentle” chemotherapy or investigational agents, treatment of infections, and supportive red cell and platelet transfusions may be more appropriate.
AML and ALL are due primarily to increased proliferation and failure of differentiation. Most leukemias arise de novo. AML may also result from evolution of myelodysplastic syndromes or progression of myeloproliferative neoplasms. Either AML or ALL may evolve from CML. Prior chemotherapy can initiate DNA damage that, if not repaired, leads to AML. If alkylating agents or radiation is implicated, the usual latency is 5–10 years and loss of chromosomes 5 or 7 is frequent. If topoisomerase II inhibitors are implicated, the usual latency is one to five years and translocations involving 11q23 are frequent. Prior chemotherapy may also lead to ALL, but rarely. Certain chemical exposures, such as benzene, are also leukemogenic. Down syndrome is characteristically associated with acute megakaryocytic leukemia, other AML subtypes, or ALL, as well as a transient myeloproliferative disorder in newborns. Familial predisposition to AML is described. Examples include kindreds with germline RUNX1 (AML1) gene mutations that are sometimes associated with thrombocytopenia and platelet function defects and kindreds with germline CEBPA mutations that are sometimes associated with increased eosinophils. Sporadic aplastic anemia and certain inherited bone marrow failure syndromes predispose to AML. Other syndromes that predispose to development of various solid tumors, such as Li Fraumeni syndrome and Bloom syndrome, also increase the risk of leukemia. In short, a family history of hematologic cancer, bone marrow failure, bleeding diathesis, and solid tumors should be sought.
The WHO classification, revised in 2008, is widely accepted but the old French-American-British (FAB) classification is still used in common parlance. The FAB classification relies upon microscopy and simple immunohistochemistry, while the WHO classification also incorporates immunophenotyping and molecular investigations.
Per the WHO, the diagnosis generally requires that very immature cells, called “blasts,” comprise at least 20% of circulating peripheral blood white cells or of nucleated bone marrow cells. There are three exceptions to this rule: (1) cases with particular recurrent, balanced translocations or inversions are considered AML even at lesser blast counts; (2) certain AML subtypes when abnormal promyelocytes, promonocytes, monocytes, or proerythroblasts count as “blast equivalents”; and (3) myeloid sarcomas, which may be unaccompanied by circulating leukemia or increased bone marrow blasts, but for all intents and purposes are a form of AML. Normal bone marrow comprises < 5% blasts and normal blood contains none.
It is often impossible to distinguish the lineage of poorly differentiated leukemia under the light microscope. Auer rods are pathognomonic of AML (Figure 179-1). In AML, immunophenotyping by flow cytometry will show typical combinations of myeloid markers (eg, CD33, CD13, CD15, HLA-DR), monocytic markers (eg, CD4, CD14, CD11b, CD11c, CD64, CD36, CD68, CD163, lysozyme), and rarely megakaryocytic or erythroid markers. In ALL, lymphoid markers are expressed (eg, TdT and CD10, as well as B cell markers CD19, cytoplasmic CD79a, cytoplasmic or surface CD22, or T cell markers CD2, CD3, CD5, CD7). Either leukemia may express markers of primitive blasts (eg, CD34, CD117). Biphenotypic and bilineage leukemias are encountered occasionally.
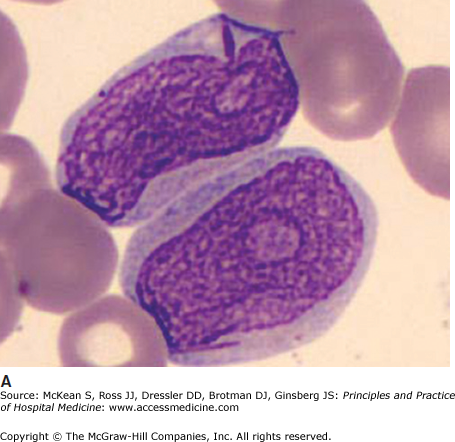
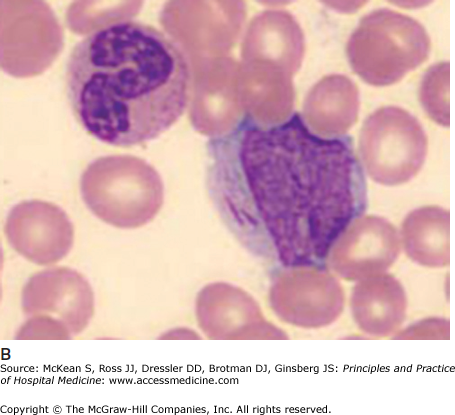
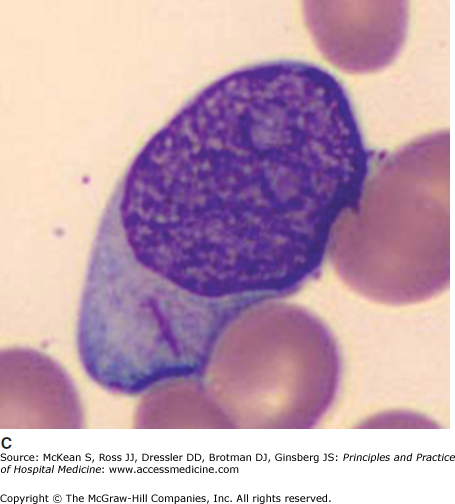
Immunohistochemistry is still useful; myeloperoxidase stains myeloblasts, nonspecific esterase stains monoblasts, and periodic acid Schiff (PAS) stains lymphoblasts. Cytogenetics or molecular analyses (FISH or PCR) may show characteristic translocations, deletions, or fusion genes. These are routine investigations. Sequencing for point mutations and internal tandem duplications of Flt3 and other genes is increasingly performed for risk stratification, and the results may steer some patients toward trials of Flt3 inhibitors or to transplant.
Blasts overtake the marrow, preventing normal hematopoiesis. Patients may come to clinical attention because of classic symptoms related to anemia, infection (due to neutropenia), bruising, or abnormal bleeding (usually with platelets < 20 × 109/L). Some patients are leukopenic, even when the marrow is “packed” with blasts, but most have normal to extremely elevated white counts. Blasts are ineffective at fighting infections. Although fever may be disease related, cultures of blood and urine should be done in all cases, and sputum and stool cultures when localizing symptoms are present. Gram-negative sepsis due to translocation of gut organisms is common when the neutrophil count is < 0.5 × 109/L. In the setting of neutropenic fever or suspected infection, broad spectrum antibiotics that cover Pseudomonas aeruginosa and other gut pathogens should be initiated promptly (eg, piperacillin/tazobactam 4.5 gram IV every 6 to 8 hours, ceftazidime 2 gram IV every 8 hours). Bone pain may occur as a result of rapid bone marrow expansion from proliferating blasts. In such cases, narcotic analgesia may be appropriate; nonsteroidal anti-inflammatory drugs and acetaminophen are usually avoided owing to increased bleeding risks (in the setting of thrombocytopenia) and masking of fevers respectively. Despite narcotics, pain relief may be suboptimal until chemotherapy takes effect, often just a few days into the treatment course.
Tissue infiltration is typical of certain AML subtypes, especially monoblastic and monocytic forms (FAB subtype M4/5) and M2 with t(8;21). Rarely, AML cells can form a solid tumor, called a myeloid sarcoma. These may be located anywhere in the body including skin, lymph nodes, gastrointestinal tract, bone, soft tissue, testes, and mediastinum. Lymphadenopathy is rarely seen with AML but is more common with ALL. Splenomegaly may occur with either. Large anterior mediastinal tumors are typical of T cell ALL. When associated with airway obstruction or the superior vena cava syndrome they are medical emergencies that should be handled with the involvement of anesthesia and intensive care teams. Prompt institution of steroids is required and chemotherapy should follow as soon as the diagnosis is confirmed. Mediastinal radiation is indicated only if the patient’s condition deteriorates and the mass grows despite these measures. Leukostasis is much more likely to occur in AML with white counts exceeding 200 × 109/L and is rare in ALL. Hyperfibrinolysis and disseminated intravascular coagulation are classically described with acute promyelocytic leukemia (M3) but can occur in other subtypes and should be routinely evaluated in all newly diagnosed patients. Markers of tumor lysis need to be checked in all patients; abnormalities detected prior to initiating chemotherapy (spontaneous tumor lysis) must be promptly treated. Patients with normal tumor lysis markers prior to chemotherapy also require treatment to prevent the development of tumor lysis syndrome, which commonly complicates the management of aggressive hematologic malignancies once chemotherapy is started (see chapter 180 for more details). Hypokalemia sometimes complicates FAB M4 or M5 AML subtypes because monocytic forms release lysozyme that damages renal tubules, causing potassium wasting. It should not be over-vigorously corrected because hyperkalemia may supervene once chemotherapy is initiated.
Central nervous system involvement is more frequent with ALL. Neurologic symptoms are usually due to leptomeningeal disease. Early CNS involvement is very rare in AML. ALL protocols all mandate a diagnostic lumbar puncture at presentation, even in the absence of neurologic symptoms. Patients with CNS disease are treated with intensified intrathecal chemotherapy and, depending on the extent of CNS involvement and the protocol, cranial irradiation. ALL patients without CNS involvement have a sufficiently high risk of future CNS disease that intrathecal chemotherapy, high dose systemic chemotherapy that crosses the blood-brain barrier, and cranial irradiation on a prophylactic schedule are routinely included in treatment protocols.
Induction therapy requires admission to the hospital. Central venous access is essential for multiagent intravenous chemotherapy. Supportive care includes prevention and management of tumor lysis syndrome (see chapter 180), treatment of neutropenic fever and infections, and administration of blood products.
For AML and ALL, the first day of chemotherapy is considered “Day 1.” A typical AML induction regimen is “3 + 7”, that is, three daily infusions of an anthracycline overlapping with seven days of continuous-infusion cytarabine, although other acceptable regimens exist. Marrow aplasia generally lasts four to six weeks. Some institutions examine the bone marrow around the fourteenth day of induction. If the bone marrow is hypoplastic and contains < 5% blasts, the patient is considered to have responded to chemotherapy. If the marrow is > 20% cellular with > 5% blasts, the response is inadequate and a second round of induction (usually with alternative drugs) may be administered. Intermediate results such as a hypoplastic marrow with > 5% blasts might prompt reexamination of the marrow in a few days’ time. Other institutions only examine the marrow once reconstitution of blood counts has occurred. The bone marrow is then examined to confirm histologic remission (ie, a normocellular marrow with < 5% blasts), although cytogenetic and molecular testing may still detect the malignant clone at this point. Patients are generally discharged after peripheral blood counts recover.
Consolidation therapy generally consists of three to four monthly cycles. Again, several acceptable regimens exist. One common regimen employs high-dose cytarabine administered every 12 hours on days 1, 3, and 5. Patients may be hospitalized for the few days of chemotherapy infusion but are usually discharged immediately thereafter. Supportive transfusions are routinely provided through the outpatient hematology-oncology clinics, but it is common for patients to be readmitted to hospital for fever/infection.
For acute promyelocytic leukemia (FAB M3), the differentiating agents ATRA and arsenic are added to induction and consolidation therapy. APL is currently the only type of AML for which maintenance therapy is employed.
Regimens for ALL are generally more complicated. They entail four phases: remission induction, intensification (also called consolidation in some protocols), continuation (also called maintenance), and CNS therapy. In adults, four anchor drugs are included in induction: glucocorticoids, vincristine, L-asparaginase, and an anthracycline. Some regimens incorporate other drugs too. The intensification phase lasts many weeks. Intensification employs multiagent chemotherapy. Continuation relies on oral daily 6-mercaptopurine and weekly oral or IV methotrexate with pulses of vincristine and steroids for one to two years or even longer in some protocols. CNS-directed treatment is given throughout all phases with intrathecal methotrexate and/or cytarabine, but an aggressive schedule of CNS prophylaxis with intrathecal drugs and/or cranial irradiation is intercalated into many protocols between segments of intensification therapy. Residual mediastinal masses are generally irradiated. Patients may be offered allogeneic stem cell transplantation in lieu of intensification or continuation therapy depending on their risk stratification. Recent studies suggest that allogeneic transplant offers a substantial survival advantage compared to chemotherapy alone in certain patient subgroups, but also that transplant-related mortality may be considerable in patients older than 35 years.
A coagulopathy related to depletion of specific proteins involved in maintenance of normal hemostasis, particularly fibrinogen and antithrombin III, can develop in patients treated with L-asparaginase. Usually this is subclinical, but bleeding and thrombotic complications occasionally develop. Patients at increased risk for bleeding complications who develop abnormalities in coagulation testing (eg, severe hypofibrinoginemia) should be considered for prophylactic factor replacement. Adults presenting with venous thrombosis should be treated with unfractionated heparin (UFH) or low molecular weight heparin (LMWH). Our general practice is to use a full-dose UFH or LMWH at platelet counts above 50 × 109/L and to consider reduced doses of UFH or LMWH at platelet counts lower than 50 × 109/L. Patients sometimes receive platelet transfusions to permit adequate anticoagulation, but this approach is not feasible for many because of eventual platelet refractoriness. For those who cannot be safely anticoagulated because of more profound thrombocytopenia or recent bleeding, a temporary inferior vena cava filter is placed. Other reasons to suspend anticoagulation are intramuscular injection of L-asparaginase and planned lumbar punctures.
The Philadelphia chromosome (BCR-ABL p190 variant) occurs in about 30% of adult ALL cases and portends a particularly poor prognosis. Appropriate candidates are offered up-front allogeneic stem cell transplant for Ph+ ALL as soon as they enter complete remission.
CNS prophylaxis is established in ALL but is not routinely required in AML. Patients with AML who should be considered for a screening LP include those with monocytic or myelomonocytic subtypes, those with t(8;21), biphenotypic leukemia, or a presenting WBC > 100 × 109/L. Some guidelines also recommend CNS surveillance in APL patients who develop hyperleukocytosis during therapy. If CSF cytology is positive, intrathecal chemotherapy is given twice a week until blasts clear, then weekly for four to six weeks.
Because several weeks of aplasia are anticipated following induction, and to a lesser extent consolidation chemotherapy, excellent supportive care is integral to patients’ survival.
Neutropenia is the central immune defect induced by antileukemic chemotherapy. Enteric Gram-negative bacteria, due to mucositis, and catheter-related Gram-positive bacteria are the most frequent causes of neutropenic sepsis. With prolonged neutropenia, opportunistic fungal infections become important. These include invasive candidal infections and pulmonary Aspergillosis. In patients with AML, viral infections and Pneumocystis jiroveci are less frequently encountered, as cell-mediated immunity is relatively preserved.
Antibiotics The use of broad-spectrum antibiotics for febrile neutropenia, even in the absence of an isolated pathogen, is imperative. Health care providers must be familiar with guidelines for the management of neutropenic fever and consultation with the infectious disease service is recommended if available/appropriate.
The use of prophylactic antibiotics is controversial. The National Comprehensive Cancer Network (NCCN) and the Infectious Disease Society of America (ISDA) state that prophylactic fluoroquinolone antibiotics may be offered to leukemia patients who are anticipated to be neutropenic (ANC < 1 × 109/L) for longer than seven days. There is evidence from placebo-controlled, randomized trials in leukemia and lymphoma and from large meta-analyses that such antibiotics dramatically decrease the frequency of fever and of documented Gram-negative infections and may decrease overall mortality. Levofloxacin may be superior to other quinolones because of its additional Gram-positive coverage. Prophylactic antibiotics are likely cost effective in high risk patients, but they may increase the frequency of Clostridium difficile infection and of Gram-positive infections like Streptococcus viridans. PCP prophylaxis is recommended for patients with ALL, but is not used in all protocols.
The use of prophylactic antifungals during induction is widely accepted. Fluconazole covers most but not all Candida species while the other antifungals mentioned below cover molds as well as yeast. In a randomized trial, prophylactic posaconazole led to fewer invasive fungal infections and reduced overall mortality compared to fluconazole or itraconazole in neutropenic AML patients receiving induction chemotherapy and the NCCN recommends its use in this setting. Many institutions continue to use fluconazole, which is less expensive and available intravenously. Also, ALL patients should receive fluconazole prophylaxis despite its narrower spectrum of activity because posaconazole, itraconazole, and voriconazole reduce clearance of vinca alkaloids, which might cause neurotoxicity. Patients with chronic severe neutropenia due to aplastic anemia or MDS should also receive prophylactic antifungal therapy. There is insufficient data to advise primary antifungal prophylaxis in consolidation therapy of leukemia.
Prophylactic growth factor support Granulocyte colony stimulating factor (G-CSF, filgrastim) is widely used in neutropenic patients with ALL. Prophylactic G-CSF enables ALL protocols in adults to be intensified, approximating the more successful pediatric protocols. However, the American Society of Clinical Oncology (ASCO) does advise caution in prescribing G-CSF to children with ALL because there is some suggestion that growth factors increase the risk of secondary MDS/AML.
Related posts:
 Strategies for Cost-Effective Care
Strategies for Cost-Effective Care
 Building, Growing, and Managing a Hospitalist Practice
Building, Growing, and Managing a Hospitalist Practice
 Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
 Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


