KEY POINTS
Primary injury occurs at the moment of trauma and is the result of direct damage to brain tissue. All subsequent preventable brain injuries are termed secondary injuries.
Head trauma is associated with cervical spine injury and stabilization of the spine (eg, cervical collar, log rolling) is maintained until the spine is cleared.
Hypoxemia, hypotension, and raised intracranial pressure (ICP) are the leading causes of death in severe traumatic brain injury (TBI) and are related to the severity of the brain injury as well as the systemic complications.
Critical care of the TBI patient is centered on airway control, favoring early intubation, resuscitation, maintenance of homeostasis, early detection of neurosurgically treated complications, and interpretation of information from bedside monitors to minimize disruption of cerebral perfusion, (oxygenation and nutrient supply) in order to prevent or limit secondary injury.
Level II evidence supports a minimum systolic blood pressure of 90 mm Hg. An SBP of <90 mm Hg must be avoided if possible, or rapidly corrected.
Currently there is no evidence from controlled clinical trials to indicate an optimal CPP goal in terms of reducing secondary ischemic injury or improving the neurological outcome; however, published guidelines state as a level III recommendation that the treatment range for CPP should be 50 to 70 mm Hg. Maintaining CPP >70 mm Hg has been associated with the development of acute respiratory distress syndrome (ARDS).
TBI is the second highest risk factor for the development of venous thromboembolism (VTE), second only to acute spinal cord injury and the incidence of deep venous thrombosis (DVT) 7 to 10 days after TBI is as high as 31.6% even with mechanical prophylaxis.
Antiseizure prophylaxis with phenytoin is recommended for the prevention of early posttraumatic seizures, that is, within 7 days of the TBI. Routine prophylaxis later than 1 week following TBI is not recommended
Recent studies have not demonstrated an overall beneficial effect of steroids on outcome and there is level I evidence that high-dose methylprednisolone increases mortality after moderate to severe TBI.
After TBI, persistent ICP >20 is associated with poor outcome and there are limited data—class III and II level evidence—that patients responding to ICP lowering treatments have a lower mortality and better outcome.
INTRODUCTION
Traumatic brain injury (TBI) is a major cause of morbidity and mortality worldwide and in the United States. TBI is caused by a blunt force or penetrating injury to the head that causes brain dysfunction. The severity of TBI may be evident immediately or may initially appear to be mild, only to deteriorate later and often rapidly. Symptoms of traumatic brain dysfunction include unconsciousness, amnesia, focal deficits, and cardiorespiratory instability due to brain stem dysfunction. TBI may be isolated but is often accompanied by additional injuries.
Of an estimated 1.7 million people in the United States that sustain TBI each year, about 52,000 die before reaching the hospital and 275,000 are hospitalized.1 TBI accounts for one-third of trauma-related mortality.1 Children less than 5 years of age, teenagers aged 15 to 19, and adults over 65 are the most likely to sustain TBI. Patients over the age of 75 have the highest rates of TBI-related hospitalization and mortality. Males, in any age group, are more likely than females to suffer TBI. Including all age groups, falls are the leading cause of TBI (35.2%), but motor vehicle crashes (MVC), the second leading cause of TBI (17.3%), are the leading cause of TBI-related death (31.8%).1 The elderly are more likely to present with fall-related TBI and young adults aged 20 to 24 years are more likely to die of MVC-related TBI. In military combat, blast injuries (61.9%) and gunshot wounds (19.5%) account for the majority of TBI.2 The rising incidence of TBI may be related to both an aging population as well as overall population expansion.1
HEAD TRAUMA: MECHANISMS OF INJURY
Head injuries can result from direct blunt or penetrating trauma to the head and from indirect processes including acceleration-deceleration and blast forces.
Direct trauma leads to scalp and skull injury and both direct and indirect injury can damage the dura, blood vessels, and brain. Penetrating trauma may result from relatively slow moving objects such as knives or projectiles such as bullets, or blast-generated fragments moving at supersonic speeds that damage the scalp, skull, and neurovascular tissue by laceration, thermal, and pressure-generated forces. Blunt or nonpenetrating injuries result in skull fracture, direct trauma to underlying neurovascular tissues and indirect injuries. Indirect injuries result from the sudden acceleration and deceleration of the brain floating within the surrounding cerebrospinal fluid (CSF), encased by the dura and rigid cranial vault, leading to rotational and shearing forces that impact neurovascular tissue against bone. TBI from blast injury may be related to indirect injury generated by pressure or shock waves and other less understood factors.
Contre-coup brain injury refers to contusions or other lesions that occur on the side contralateral or 180° from the force of impact; coup injury refers to ipsilateral injury directly below the impact. Contusions are localized injuries to the cerebral parenchyma that occur when the brain is pushed or jarred against the bony components of the skull resulting in hemorrhage, edema, or necrosis. Contusions are typically observed at the frontal poles, orbital frontal lobes, temporal poles, and cortex above the Sylvian fissure3 (Fig. 118-1).
Skull fractures may be single or multiple, linear, or depressed (Fig. 118-2). Basilar fractures are associated with cerebrospinal fluid (CSF) leak and meningitis as well as a greater risk of cranial nerve and vascular injury. Scalp lacerations above fractures are termed open fractures and have a greater risk of infection.
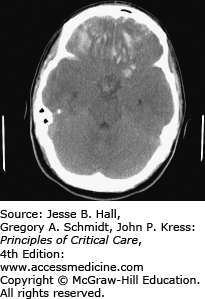
Temporal bone fractures can injure the middle meningeal artery or a branch thereof resulting in hemorrhage into the epidural space, between the inner table of the skull and above the dura termed epidural hematoma. Epidural hematomas may also result from meningeal vein or dural sinus damage. Epidural hematomas are more common in children and young adults since the dura is not as adherent to the skull as in the elderly (Fig. 118-3). The classic clinical presentation of epidural hematoma is a brief loss of consciousness followed by a neurologically intact interval, followed by sudden deterioration, coma, and death from herniation within hours. The treatment is immediate neurosurgical evacuation or, if not available in time, a burr hole can be lifesaving.
FIGURE 118-3
Head CT revealing a large, fatal, epidural hematoma (EH) in a patient who fell while intoxicated. Note the lenticular shape which is characteristic of EH. EHs are usually limited by sutures where the dura is most adherent. The anterior portion is limited by the coronal suture, while the posterior portion is limited by the lambdoid suture. EHs are easily treated with surgery and are usually associated with an excellent prognosis if recognized early.
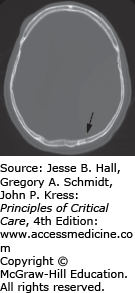
Subdural hematomas collect beneath the dura and result from laceration of the bridging cortical veins. The venous bleeding that results in subdural hematomas usually is slower, resulting in a more gradual decline in consciousness, but when they become large, rapid evacuation is needed. They are typically seen in older patients usually occurring after falls, assaults, or MVAs and are associated with greater underlying brain damage than extradural hematomas (Fig. 118-4).
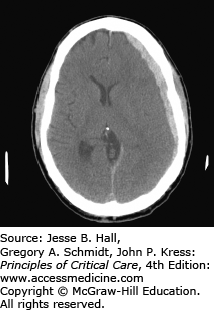
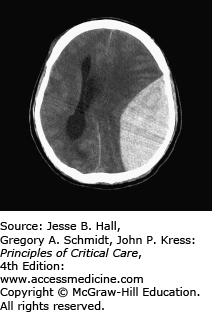
Trauma is the most common cause of subarachnoid hemorrhage (SAH) (Fig. 118-5). The incidence of SAH associated with head trauma has been reported to range from 33% to 60%.4 Traumatic subarachnoid, intraventricular, and intraparenchymal hemorrhages are associated with greater degrees of injury and poorer outcomes. Intraparenchymal hemorrhages may be focal or multifocal and are less frequent than epidural or subdural hematomas in nonpenetrating injury (Fig. 118-6). Traumatic subarachnoid hemorrhage, like that due to ruptured aneurysms, can lead to vasospasm, and occasionally ischemic deficits. Transcranial Doppler sonography is useful in detecting vasospasm.
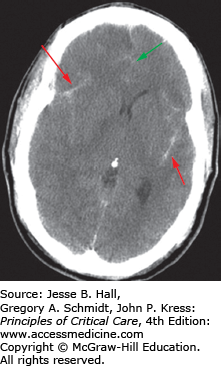
FIGURE 118-6
CT scan showing a focal right frontoparietal intraparenchymal hematoma (red arrow) associated with a subdural hematoma. There is shifting of the midline to the left due to mass effect from the hematomas (green arrow). This patient ultimately required a decompressive craniectomy for elevated intracranial pressure.
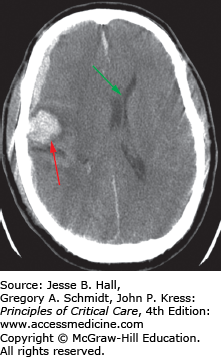
Brain swelling with associated intracranial hypertension develops in 10% to 15% of severe TBI patients who have an initial normal head CT, and in 53% to 63% of patients with acute traumatic abnormalities on a hospital admission CT.5 Expansion of the intracranial components, for example, brain edema or hematoma, leads to increased ICP that can lead to herniation.
Expansion of the temporal lobe can lead to transtentorial uncal herniation with compression of the ipsilateral third cranial nerve containing peripheral parasympathetic nerve fibers and ipsilateral cerebral peduncle compression resulting in the classic findings of an ipsilateral dilated and fixed pupil with contralateral hemiparesis. Less commonly, if the brain stem is displaced against the tentorium on the side opposite to the herniation, hemiparesis or hemiplegia can occur on the side ipsilateral to the herniation, the so-called “Kernohan notch” phenomenon. In central transtentorial herniation, the thalamic area is displaced over the tentorial notch leading to decorticate posturing and possibly rupture of the paramedian branches of the basilar artery causing “Duret” hemorrhages. Herniation of the cerebellar tonsils through the foramen magnum or tonsillar herniation can result in cardiorespiratory arrest from compression of the medulla. Transcalvarial herniation may occur through a skull fracture and subfalcine herniation occurs when the cerebrum herniates below the falx cerebri due to midline shift (Fig. 118-6) resulting in possible compression of the anterior cerebral artery.
TBI may result in focal and/or diffuse lesions. Neuronal cell bodies may be damaged leading to focal gray matter ischemia, necrosis, and focal deficits. The shearing of axons in the cerebral white matter due to the differential acceleration of gray versus white matter tissue of different densities, may lead to axonal degeneration, termed diffuse axonal injury (DAI), with nonfocal neurologic deficits such as encephalopathy and coma (Fig. 118-7). DAI most often occurs in the setting of rapid, high magnitude acceleration or deceleration. DAI occurs in more than 50% of all severe head trauma and in more than 85% of the subset related to motor vehicle collisions.5 Deficits in memory and learning are common after TBI and may be related to frontal lesions.6
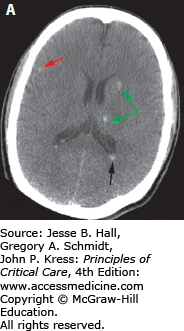
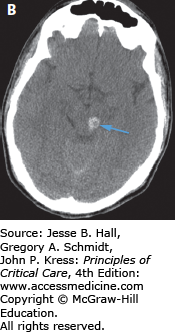
FIGURE 118-7
This patient was a pedestrian hit by a car. He suffered severe diffuse axonal injury (DAI). DAI may present with essentially normal imaging that is disproportionate to the severity of the clinical examination. However, this patient exhibited typical DAI findings on CT. A. Corticomedullary junction (red arrows) and deep white matter and basal ganglia (green arrow) hemorrhage. There is also blood within the occipital horns of the ventricle (black arrow). B. The patient also exhibited a midbrain hemorrhage (blue arrow). Deep or midline hemorrhages such as those in the ventricle and corpus callosum are often indicative of severe brain disruption due to shearing forces.
PRIMARY AND SECONDARY TBI
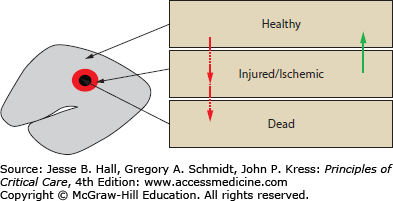
A conceptual framework with which to care for neurologically injured patients is based on the classification of injury that occurs immediately after any insult, called primary injury, and all subsequent injuries, termed secondary injuries (Fig. 118-8). In TBI, primary injury occurs at the moment of trauma and is the result of direct damage to brain tissue. After the primary injury, the remaining brain tissue consists of healthy tissue, injured (or ischemic) tissue, and dead tissue. All subsequent brain injuries are termed secondary injuries and will result in further neuronal injuries and death over hours to days after the primary injury. Secondary injuries may be caused by brain edema, hematoma expansion or delayed hemorrhage, intracranial hypertension, herniation, hypotension, hypoxemia, hypercarbia or hypocarbia, circulatory or respiratory arrest, seizures, vasospasm, and severe electrolyte disturbances. The pathophysiology of secondary TBI involves impaired cerebrovascular autoregulation, cellular metabolic dysfunction, and inadequate cerebral oxygenation.
FIGURE 118-8
Conceptual model of brain injury. Immediately after TBI or any acute neurological insult, there are damaged (dead or injured) brain cells (red arrows) and uninjured or “healthy” brain cells (primary injury). The central goal of care after TBI is the prevention of additional brain cell injury or death (secondary injury) (red arrows) and the maintenance of an environment maximally conducive to recovery of the potentially salvageable injured cells (green arrow).
On the cellular and molecular level, secondary injury results from lactic acid production and depletion of ATP due to anaerobic glycolysis, increased membrane permeability due to ion pump failure, and activation of voltage dependent calcium and sodium channels resulting in influx of calcium leading to activation of catabolic enzymes and free radical formation that leads to progressive damage to both intracellular and nuclear structures leading to membrane failure, cytotoxic brain edema, necrosis, and apoptosis.7 The release of excitotoxic substances including the amino acids glutamate and aspartate damages adjacent neurons leading to further injury. Damage to the endothelial layer of the blood brain barrier leads to vasogenic edema as well,7 but cytotoxic edema, which does not respond to steroids, is more important after TBI. Primary as well as secondary injury results in the release of proinflammatory cytokines such as tumor necrosis factor, interleukin-1-β, and interleukin-6; prostaglandins, leukotrienes, and activation of complement and coagulation systems, neutrophils, macrophages, and lymphocytes that lead to further endothelial damage and up regulation of cellular adhesion molecules such as P-selectin, intercellular adhesion molecules (ICAM-1), and vascular adhesion molecules (VCAM-1) that further facilitate the influx of leukocytes into tissues leading to further secondary brain damage.7
Cellular necrosis occurs as the result of severe mechanical and ischemia-hypoxia-induced injury. Apoptosis or programmed cell death may occur in cells that initially appear structurally intact and have adequate ATP and membrane potentials. Over hours to days after the injury, an imbalance between pro- and antiapoptotic proteins with consecutive activation and deactivation of caspases representing specific proteases of the interleukin-converting enzyme family are felt to be the most important mediators of apoptosis.7
The prevention of primary head injury is a major public health concern. Neurogenesis, the regeneration of neurons, occurs at an insignificant rate in humans8 and gene therapy is in the early research phase without clinical application.9 The prevention of secondary injury is the major goal in the treatment and optimization of outcomes of the patient after head injury. The optimal critical care of the head injured patient requires the provision and maintenance of a homeostatic environment that leads to recovery of potentially salvageable injured or ischemic brain and prevents or mitigates insults that would render injured cells dead.
INITIAL STABILIZATION, IMAGING, AND MANAGEMENT
A moderate to severely head injured patient is a trauma patient with rapid triage and stabilization beginning in the field and transport, ideally to a neurotrauma center (or the most appropriate hospital within range), facilitated by emergency medical personnel, to continued evaluation and stabilization in the emergency department utilizing the ATLS protocol by trauma surgeons and neurosurgeons, followed by transport to radiology for diagnostic imaging or operating room for acute decompression of intracranial mass lesions, or ICU—the order of which is determined by the nature of the acute injuries. Head trauma associated with cervical spine injury and stabilization of the spine (eg, cervical collar) is maintained until the spine is cleared (see Chap. 119, Spinal Injuries). Patients are admitted to the ICU depending on the risk or development of respiratory or circulatory failure, organ failure, and shock, and the severity of brain injury requiring close monitoring.
After initial stabilization, patients routinely undergo computed tomography (CT) imaging which provides immediate information regarding the presence or absence of skull and spinal fractures, foreign objects, contusions, extracranial and intracranial hemorrhages, edema, hydrocephalus, and herniation. Neurological examination may be limited by depressed consciousness. Repeat or serial CT scans are useful to determine the etiology of acute deterioration or failure to improve and to assess changes in initial lesions. When clinical deficits are not explained by CT findings, magnetic resonance imaging (MRI) is more sensitive to assess the degree of DAI which is inferred by the presence of punctuate white matter hemorrhages, but such findings may be absent on both CT and MRI.10 CT scanning is the primary study in patients with penetrating head trauma associated with metallic foreign bodies where MRI scanning is contraindicated. Multidetector CT scanning (MDCT) allows three-dimensional imaging that may assist in preoperative preparation.10 CSF leaks can present as CSF otorrhea or rhinorrhea and can be diagnosed with nuclear or CT cisternography, especially if the possible source is not clear on initial imaging. Cisternography performed after the injection of intrathecal contrast is more specific in identifying the anatomical location of the leak, while nuclear cisternography is more sensitive to the existence of the leak but not its precise location.
Upon admission to the ICU, a tertiary head-to-toe examination is performed to identify any potentially missed traumatic injuries11 and assess the current neurological exam noting any change that has occurred since the last described exam. The head is examined for ecchymoses, lacerations, deformities, signs of basilar skull fracture (raccoon eyes, Battle sign), or CSF leak (rhinorrhea or otorrhea). Neurological examination is focused on assessment of the overall mental status, cranial nerves III and VI, pupillary responses, oculocephalic (doll’s eyes), corneal and gag reflexes, deep tendon reflexes, any a symmetry or focality of extremity movement, the presence of pathologic reflexes (eg, Babinski sign or decorticate or decerebrate posturing), and sensation. Bilateral and dilated fixed pupils indicate brain stem injury; unilateral or bilateral dilated fixed pupils may occur with cerebral herniation, as does decerebrate posturing. Hypoxemia, shock, and hypothermia may cause pupillary dilation and abnormal pupillary responses. Papilledema is usually not immediately seen in acute TBI with intracranial hypertension (IH) and is a later finding.12 A negative neurologic exam does not rule out significant intracranial injuries and intracranial hematomas may be delayed, occurring days to weeks after the initial insult.
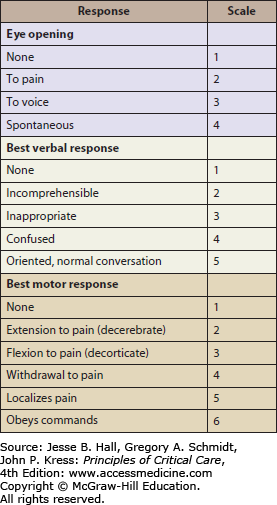
The Glasgow Coma Scale13 (GCS) (Fig. 118-9) can be used to determine the severity of head injury, for serial assessment, and has prognostic implications. It is based on the best eye opening response, verbal response, and motor response. A score of ≤8 indicates severe TBI; however, it is important to rely on the overall clinical picture, particularly when the GCS is in the mild and moderate range (9-15). Cushing reflex—bradycardia, hypertension, and apneic breathing—is a “classic” if not late sign of elevated ICP leading to cerebral herniation and terminal brain stem compression; however, acute severe hypoxemia, which can cause both hypertension and bradycardia, also requires rapid recognition and management.
Basic initial ICU monitoring and access includes continuous ECG, blood pressure via arterial line, pulse oximetry, central venous access, nasogastric tube insertion, and Foley catheter placement, if there are no contraindications. TBI is a classic risk factor for stress ulcers (Cushing ulcer14) and prophylaxis with H2-blockers should be initiated. Severe TBI is also a strong risk factor for venous thromboembolism (VTE); however, due to the early bleeding risks, mechanical prophylaxis (intermittent pneumatic compression devices) is used initially with pharmacologic prophylaxis added when the risk of bleeding has sufficiently decreased.
Additional hemodynamic and neurological monitoring depends on the clinical diagnosis and condition. Laboratory studies, either as initial or follow-up, include arterial blood gases, electrolytes, glucose, lactate, complete blood count, coagulation profile, type and cross, and liver function tests. If appropriate and omitted thus far, a toxicology screen should be ordered. Health care proxy or available family or friends should be asked to provide preaccident and accident history as well as advanced directives. A review of the diagnostic imaging, laboratory results, and surgical procedures performed thus far, and communication with the neurosurgeon regarding anticipated diagnostic imaging, neurosurgical interventions, neuromonitoring, and ICU management is essential.
Hypoxemia, hypotension, and raised ICP are the leading causes of death in severe TBI and are related to the severity of the brain injury as well as the systemic complications. Critical care of the TBI patient is centered on airway control, favoring early intubation, resuscitation, maintenance of homeostasis, early detection of neurosurgically treated complications, and interpretation of information from bedside monitors to minimize disruption of cerebral perfusion, oxygenation, and nutrient supply in order to prevent or limit secondary injury.
RESPIRATORY MANAGEMENT
Hypoxemia and hypotension are the two most important factors associated with adverse outcomes in patients after TBI, and the association with TBI is stronger than in trauma patients without neurological injury.15,16 Patients who have severe brain injury are at increased risk for acute respiratory distress syndrome (ARDS).17 Patients with severe head injury (GCS ≤8) may have an abnormal lung elasticity and resistance as early as day 1 post injury.18 A recent retrospective cohort study of the Nationwide Inpatient Sample (NIS) database reported a 22% prevalence of ARDS/acute lung injury (ALI) after TBI in 2008 with an in-hospital ARDS/ALI-related mortality of 28%.19
Hypoxemia may be caused by noncardiogenic pulmonary edema from ARDS due to a systemic inflammatory response to trauma or fat emboli, neurogenic pulmonary edema, or less commonly, cardiogenic pulmonary edema. Other etiologies of hypoxemia include airway obstruction, lung contusion from direct chest trauma, flail chest, pneumothorax, retained secretions or aspiration, pneumonia, and hypercarbia. Hypercarbia may be caused by depressed respirations from coma or brain stem dysfunction, chest trauma, airway obstruction, or high cervical spine injuries.
Oxygenation should be monitored by pulse oximetry and checked by arterial blood gases. Hypoxemia defined as [Math Processing Error] <60 mm Hg or hemoglobin-oxygen saturation <90% must be avoided.15 After TBI, patients with any of the following: signs of respiratory distress, intracranial hypertension, impending herniation, encephalopathy or coma (GCS ≤9), requiring high levels of inspired oxygen to maintain [Math Processing Error] above 60 mm Hg, absolute CO2 retention, or CO2 retention relative to respiratory minute volume should be immediately intubated. Early intubation after moderate to severe TBI is preferred to avoid the hypoxemia, aspiration, potential triggering of seizures and exacerbation of intracranial hypertension that occurs in the crashing, emergently intubated TBI patient. Endotracheal intubation and mechanical ventilation also allow therapeutic hyperventilation for temporary relief of impending herniation, procedures requiring sedation, and if necessary, pharmacologic coma.
In critically ill patients in general, and in TBI patients in particular, endotracheal intubation is significantly more difficult due to the need for precautionary neck stabilization, encephalopathy, potential for intracranial hypertension, bleeding, vomiting, copious oropharyngeal secretions, airway edema, respiratory dysfunction, and hemodynamic instability. Complications such as hypoxemia, aspiration, bradycardia, and cardiac arrest increase significantly20 as the number of laryngoscopic intubation attempts increase. Indirect optical laryngoscopy does not require aligning the head and neck and provides better visualization of the vocal cords facilitating faster, less traumatic intubation requiring less sedation and less training to become proficient compared to direct laryngoscopy.21,22
Specific considerations in TBI patients are precautionary manual in-line neck stabilization in the setting of potential acute cervical injury (see Chap. 119, Spinal Injuries) and rapid sequence intubation using sedatives and succinylcholine, a short acting paralytic agent, to avoid exacerbations in intracranial pressure.
After endotracheal intubation, mechanical ventilation should be set to an assist-control type mode with the respiratory rate and tidal volume adjusted to maintain the desired [Math Processing Error] level. The [Math Processing Error] and positive end expired pressure (PEEP) should be minimized to maintain the [Math Processing Error] >60 and the [Math Processing Error] >90. PEEP, especially in the setting of reduced pulmonary compliance, does not significantly raise the intracranial pressure.23,24 As such, PEEP does not have to be avoided if needed to maintain adequate [Math Processing Error] at less toxic [Math Processing Error] levels. Data indicate that a low tidal volume approach may be applied safely in patients who have acute intracranial disorders25; however, the significance of ventilator-induced lung injury in patients with TBI is unclear. In the setting of ARDS in the TBI patient, it is safe to institute lung-protective mechanical ventilation by reducing tidal volumes to lower plateau pressures; however, the respiratory rate should be increased to avoid acute elevations in [Math Processing Error] or frank hypercapnia that can exacerbate or result in intracranial hypertension. In patients with impending herniation or severe ICP elevation, acute hypercapnia must be avoided.
Mechanical ventilation causes positive intrathoracic pressure and higher pressures can cause decreased venous return and a rise in jugular venous pressure leading to an increase in cerebral blood volume (CBV) and in ICP and to a drop in cardiac output and blood pressure, thereby reducing cerebral perfusion pressure (CPP) and cerebral blood flow (CBF). In areas where cerebral autoregulation is intact, decreases in CPP are compensated for by cerebral vasodilation, increasing CBV and potentially increasing ICP; if autoregulation is impaired, decreased CPP may lead to cerebral ischemia. The effect of these changes on the brain is difficult to impossible to monitor, but avoiding extremes and maintaining homeostasis is critical. In the TBI patient, premature extubation may result in 2nd injury.26
Tracheostomy either by open or percutaneous dilational techniques, depending on patient anatomy and local expertise, should be performed in patients expected to require mechanical ventilation for greater than 10 to 14 days. The exact timing of tracheostomy remains a matter of debate. Tracheostomy may decrease the number of ventilator days, but there is no evidence that it decreases ICU length of stay or pneumonia rates.27,28 The benefits of tracheostomy include better oral care, improved patient comfort, decreased self-extubation risk, allowance for less sedation, better communication (speaking valve), more aggressive weaning attempts, decreased dead space ventilation, and possibly a lower work of breathing.
HEMODYNAMIC MONITORING AND MANAGEMENT
Both hypotension and raised ICP are the leading causes of death in severe TBI and are related to the severity of the brain injury as well as the systemic complications. Hypotension exclusively from TBI is a terminal event due to herniation.
Both mortality rate and outcome (ie, degree of disability) are significantly increased in patients with documented episodes of hypoxemia16 or hypotension.29 An analysis of the large, prospectively collected, observational data set, the Traumatic Coma Data Bank (TCDB), found that hypoxia and hypotension were independently associated with significant increases in morbidity and mortality in the setting of severe head injury.30 A single prehospital episode of systolic blood pressure <90 mm Hg is associated with increased morbidity and a doubling of mortality compared with a matched group of patients without hypotension.30 Hypotension is among the five most powerful predictors of outcome after TBI, independent of the other major predictors of outcome including age, admission GCS score, admission GCS motor score, intracranial diagnosis, and pupillary status.30 In the hospital, repeated episodes of hypotension and increased total duration of hypotensive episodes were significant predictors of both mortality and poor neurological outcome.31 Patients that respond to resuscitation after TBI with improved BP have a better survival.32
Hemodynamic management should employ fluids, vasoactive agents, and blood transfusions as indicated to maintain a systolic blood pressure above 90 mm Hg. The 90 mm Hg systolic pressure threshold is derived from statistical distributions of blood pressure for normal adults. Systolic blood pressures lack a consistent relationship with mean arterial pressure (MAP) and MAP is used to calculate the cerebral perfusion pressure (CPP). It may be desirable to maintain MAP considerably above those represented by systolic pressures of 90 mm Hg, but there are no studies performed to date that support a particular target blood pressure. An analysis of the relationship between admission SBP and MAP after TBI and GOS33 (Fig. 118-10) at 6 months using the IMPACT database found that SBP on the order of 135 mm Hg and MAP on the order of 90 mm Hg were associated with the best outcome,34 although these data do not support a strong causal inference. However, because of ethical considerations, there are no class I studies (ie, well-designed randomized controlled trials) of the effect of blood pressure resuscitation targets on outcome.15 As such, level II evidence35 (Fig. 118-11) supports a threshold systolic blood pressure of 90 mm Hg.15 A SBP of less than 90 mm Hg must be avoided if possible, or rapidly corrected.
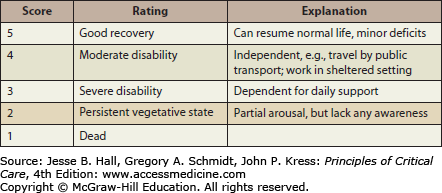
FIGURE 118-11
Brain Trauma Foundation Evidence Levels for TBI Recommendations.35 (Data from Carney NA. Guidelines for the management of severe traumatic brain injury. Methods, J Neurotrauma. 2007;(24 suppl 1):S3-S6).

The type of hemodynamic monitoring employed should be determined by the severity of TBI, the degree of instability, the response or lack of response to resuscitation, and the expertise of the critical care physician. Foremost, no matter what type of monitoring is employed it must be coupled with the clinical context including physical examination, intake and output, pertinent labs including hemoglobin, renal function, lactate, ABG, CXR, ECG, CT (intracranial pathology), ICP, and the results of any additional neuromonitors, for example, brain tissue oxygen, CBF, etc.
Hemodynamically stable patients may be monitored simply by continuous blood pressure via arterial catheter and ECG. In patients with severe or persistent hypotension, shock, multiple organ dysfunction, and intracranial hypertension, the titration of fluid and vasoactive agents is more challenging. Vasopressors in the setting of intravascular volume depletion may worsen cerebral ischemia and other organ perfusion and excess fluid resuscitation may lead to worsening pulmonary edema, hypoxemia, cerebral ischemia, and cerebral edema.
Central venous pressure (CVP) traditionally has been used to assess the adequacy of intravascular volume and although it is still often measured and discussed in neurosurgical ICU settings, it should not be used to guide fluid management.36 Recent studies have failed to demonstrate a clinically useful correlation between absolute CVP or change in CVP with intravascular volume or right ventricular preload,37 and it does not predict fluid responsiveness.36 The pulmonary artery occlusion pressure obtained from the pulmonary artery catheter (PAC) also does not provide accurate information about left ventricular preload or intravascular volume.37
Recent trends in hemodynamic monitoring in the critically ill favor the use of less invasive and more direct measures of cardiac function38,39 and dynamic indices predictive of preload (fluid) responsiveness.40 Bedside echocardiography can rapidly and directly assess both right and left ventricular preload and contractility and in trauma patients can rule out significant pericardial effusion.39 The lungs can be assessed by ultrasound on the same examination, to detect pulmonary edema (B-lines) early on, rapidly rule out a pneumothorax (presence of lung sliding) or hemothorax, and can detect atelectasis (shift of heart, echogenic lung appearance).39 Ultrasound can also be used to determine the inferior vena cava (IVC) diameter and variability with respiration as a dynamic index of fluid responsiveness,40 although it may be inaccurate in the setting of intra-abdominal hypertension, RV dysfunction, or pericardial tamponade. Pulse contour analysis of the arterial pressure waveform can measure the pulse pressure variability with respiration. Values greater than 12% to 13% are more predictive of fluid responsiveness41; however, the cardiac rhythm must be sinus, the tidal volumes constant and adequate without spontaneous respiratory efforts—and this requires a heavily sedated or paralyzed patient on continuous mechanical ventilation. Increases in cardiac output (CO) in response to passive leg raising (PLR) have been proposed to determine preload responsiveness regardless of respirations or arrhythmias, but whether the effects of PLR are due to volume (autotransfusion of blood) versus sympathetic stimulation from PLR is not entirely clear. Regardless of these issues, it would be prudent to avoid PLR in patients with increased intracranial pressure.
Serial measurements of CO are more technically difficult with echocardiography. Pulse contour analysis can provide a continuous cardiac output and relative trends in the cardiac output, but are subject to changes in the arterial pressure waveform not necessarily related to changes in CO and do not provide an accurate absolute cardiac output compared to thermodilution techniques. Some manufactures allow or require calibration of the pulse contour CO with transpulmonary lithium or transpulmonary thermodilution dilution techniques. These provide accurate CO, but within 60 minutes the pulse contour-derived CO drifts beyond the 30% error range compared to thermodilution and the TD CO must be repeated if an absolute CO is needed.42
Due to a lack of evidence across multiple studies that PAC monitoring improves the outcome, use of the PAC has decreased significantly in the ICU. However, for the intensivist experienced in its insertion and data interpretation, the PAC can provide accurate pulmonary artery pressures, right heart thermodilution cardiac output, and true mixed venous blood gases. The venous oxygen level can be misleadingly normal in the face of regional hypoperfusion and does not correlate with cardiac output. The central or mixed venous carbon dioxide (CO2) levels and the venous-arterial CO2 difference correlate better with perfusion and cardiac output43 and if elevated may indicate a low cardiac output, hypermetabolic state, or ongoing regional hypoperfusion.
There is no particular target cardiac output or index number for patients with TBI (or any other critical illness); however, when there is evidence of hypoperfusion and the CO may be inadequate, measures to increase the CO by fluid resuscitation and inotropes may be instituted.
To date, there is a lack of clinical data on the effect of changes in CO on cerebral perfusion and the studies have focused primarily on blood pressure with the goal of maintaining SBP at least above 90 mm Hg. Currently the most rational approach appears to be maintaining homeostasis and not driving hemodynamics toward arbitrary end points. Normal or adequate parameters of pressure and cardiac output are preferable to maximization strategies that may result in further organ dysfunction and hence, cerebral ischemia. Maintaining adequate intravascular volume, blood pressure, and cardiac output—which is not necessarily monitored but can be inferred by adequate urinary output, normal or decreasing lactate, and physical signs of adequate perfusion—is recommended. Choices of fluid and vasoactive agents should be based on the patient’s current cardiac, pulmonary, and renal function, assessment of intravascular volume status, presence of SIRS or sepsis, presence of cerebral edema, intracranial hypertension, the results of monitors of cerebral oxygenation or perfusion, and the pharmacological actions of the vasoactive agents. The decision to monitor cardiac output is made when the clinical signs are not correlating and there are questions about the response to therapy.
Advanced neuromonitoring techniques increasingly allow improved assessment of the effects of changes in systemic hemodynamics on the brain, but in the absence of defined protocols that clearly improve outcome, maintaining normal homeostatic parameters may be the optimal approach.
PAROXYSMAL SYMPATHETIC HYPERACTIVITY
Dysautonomia, or the more recently applied term, paroxysmal sympathetic hyperactivity (PSH), occurs in approximately 7.7% to 33% of patients with severe TBI admitted to the intensive care unit. PSH can be transient or prolonged and is characterized by tachycardia, tachypnea, hypertension, hyperthermia, diaphoresis, pupillary dilation, abnormal posturing, and hypertonia. The etiology remains unclear, but may represent a dissociation of the brain stem from higher sympathetic regulation or control. PSH has been managed with β-antagonists, such as propranolol, benzodiazepines, gabapentin, bromocriptine, and intrathecal baclofen.44
INTRAVENOUS FLUID AND ELECTROLYTE MANAGEMENT
The type and volume of intravenous fluids utilized after TBI are based on the objectives—providing maintenance fluid, volume resuscitation, treatment of hypernatremia or hyponatremia, and treatment of intracranial hypertension—and are modified based on systemic hemodynamics (see above), serum sodium levels, renal function, and presence of post-TBI posterior pituitary gland dysfunction.
Sodium disorders are common after TBI with greater incidence reported in patients with SDH, intracerebral hematoma and DAI.45 Hypernatremia is associated with a higher mortality after moderate to severe TBI likely reflecting the severity of brain injury46 and although hyponatremia has not been clearly linked to mortality after TBI, the presence of even mild hyponatremia on general hospital admissions is associated with increased mortality.47 TBI can cause injury to the pituitary gland and hypothalamic tracts (edema, direct damage) resulting in central diabetes insipidus (DI) and hypernatremia or the syndrome of inappropriate antidiuretic hormone (SIADH) and hyponatremia.48
Causes of hypernatremia post-TBI include DI, hypernatremic fluid administration, and hyperosmolar therapy. DI usually presents with polyuria whether immediately after TBI or within the first 2 to 3 days.49 The diagnosis of DI is supported by polyuria in the absence of confounding causes such as osmotic diuresis (eg, hyperglycemia, mannitol), hypernatremia, and hypotonic urine with urine osmolality less than serum osmolality. DI is treated with desmopressin (1-desamino-8-D-arginine vasopressin [DDAVP]) under close monitoring of fluid intake, output, and serum sodium levels. DI may be transient so that prn dosing is preferred initially; if DI persists beyond 2 days a regular dosing regimen is used.49 Occasionally, SIADH may manifest after initial DI and rarely DI may return permanently after SIADH—called a “triple phase response.”49 SIADH in this case may be due to the release of stored arginine vasopressin (AVP) from damaged neurons followed by a lack of AVP if insufficient functional neurons remain. Therefore, periodic reassessment for SIADH and DI is wise. Both DI and SIADH usually occur acutely within the first week post-TBI and SIADH usually resolves within 6 months, but rarely DI may persist.48
The most common etiology of hyponatremia post-TBI is SIADH, which accounts for 80% of cases.49 Brain tissue injury, elevated ICP, extracranial trauma, and surgery are factors that may lead to inappropriately elevated AVP. Another proposed mechanism for post-TBI hyponatremia is termed cerebral salt wasting (CSW) or renal salt wasting,50 caused by natriuretic peptide release from injured brain tissue leading to natriuresis and hypovolemia. ACTH deficiency occurs acutely in about 15% of patients post-TBI; however, hyponatremia is only rarely due to glucocorticoid deficiency.51
The differentiation between SIADH and CSW is not straightforward. There is a complete overlap in laboratory parameters (including urine sodium and osmolality) with both conditions resulting in hyponatremia and natriuresis. The diagnosis hinges upon an accurate assessment of volume status; however, the traditional reliance on CVP leads to frequent errors in intravascular volume assessment.36 Although controversy exists about the existence of CSW and it may be relatively rare compared to SIADH,49 euvolemia is consistent with SIADH and is treated with fluid restriction while hypovolemia is consistent with CSW that responds to saline hydration. There is a risk if fluid restriction is applied to the TBI patient with CSW as this can exacerbate hypovolemia and potentially compromise cerebral perfusion. In the TBI patient with hyponatremia, it is important to assess the serial changes in body weight and cumulative fluid balance up to that point coupled with a reliable evaluation of hemodynamics (ie, not relying on CVP measurements—see previous section “Hemodynamic Monitoring and Management”).
Both continuous maintenance fluids and fluid boluses for resuscitation should be provided with the goal of maintaining a euvolemic state while avoiding hypovolemia. Both hyper- and hypovolemia are associated with worse outcomes after TBI.52 Isotonic fluids that help to maintain the intravascular volume and not exacerbate cerebral edema or the tendency toward hyponatremia, such as normal saline are employed for both continuous maintenance and bolus resuscitation. If hyperchloremic metabolic acidosis occurs, a balanced isotonic fluid with buffer capacity (eg, acetate) such as Plasmalyte may be used. Plasmalyte also contains magnesium and potassium which is convenient since hypomagnesemia and hypokalemia are common after severe TBI as is hypophosphatemia and all of these electrolytes frequently require supplementation.53 Although hypotonic fluids such as ½ normal saline or 5% dextrose in water are usually avoided, they may be indicated if there is significant hypernatremia, for example, >155 mEq/L. Severe hypo- or hypernatremia should be corrected gradually to avoid rapid fluid shifts across the blood brain barrier than can result in central pontine myelinolysis or cerebral edema.
We have instituted the use of a continuous 3% saline infusion in moderate and severe acute TBI patients upon admission in order to prevent potentially harmful and commonly seen posttraumatic hyponatremia that can cause significant brain swelling. The therapeutic goal is maintenance of serum sodium levels at 140 to 145 mEq/L. Although the effect on outcome after TBI has not yet been determined, it does appear to be successful in avoiding severe hyponatremia while helping to maintain intravascular volume regardless of the etiology—SIADH or CSW (see also “Hypertonic Saline” in “Treatment of Intracranial Hypertension”).
BLEEDING AND TRANSFUSION ISSUES
Acute blood loss anemia is never primarily caused by closed space intracranial bleeding since death from herniation would occur far sooner than anemia. Acute blood loss anemia, decreased red cell production, and coagulopathies occur as the result of traumatic injuries dictated by the degree of multisystem trauma. The concern after TBI is maintaining perfusion and oxygen delivery to the brain to prevent further secondary injury and hypotensive anemia should be treated with blood transfusions while the source of bleeding is determined and controlled. There is no evidence to define a particular target hemoglobin, platelet transfusion, or INR threshold after general trauma, including after TBI. An analysis of the transfusion requirements in critical care (TRICC) trial subset of multitrauma patients suggests that a restrictive transfusion target of Hb 7 g/dL is not inferior to a liberal target Hb of 10 g/dL.54 However, the transfusion threshold after TBI remains controversial. A retrospective review of patients with severe (GCS <8) isolated TBI concluded that a restrictive transfusion practice (blood transfusion trigger Hb <8 g/dL vs Hb 8-10 g/dL range) is safe.55 A recent retrospective review of 139 patients who were admitted with TBI and moderate anemia (hematocrit 21-30) found no association between blood transfusion and mortality, but blood transfusions and the transfusion volume were associated with poorer long-term functional outcomes.56 The authors concluded that transfusion should be aimed at patients with symptomatic anemia or physiological compromise.56 Another study hypothesized that blood transfusions resulting in hematocrit values >28% at the end of the initial operating room phase would result in more complications, increased mortality, and impaired recovery in severe TBI patients,57 139 TBI cases were retrospectively reviewed and blood transfusion resulting in hematocrit values >28% was not associated with improved or worsened outcome.
Coagulopathy after general trauma is not well studied.58 Traumatic coagulopathy can be explained at least in part by tissue factor release into the general circulation with activation of the coagulation cascade in both TBI and non-TBI.59 After TBI, abnormalities in coagulation include disseminated intravascular coagulation (DIC) (triggered by systemic inflammation and tissue thromboplastin release or with multisystem trauma consumptive coagulopathy due to bleeding), thrombocytopenia, elevated INR, PTT, and hypofibrinogenemia. DIC occurs in 8% to 76% of patients after TBI depending on the definition of DIC and the severity of TBI.60 Consumptive coagulopathy due to bleeding requires surgical control of the bleeding source. Fresh frozen plasma with target INR below 2.0 and platelet transfusions to keep the levels at or above 50,000/mm3 in the acute phase, if there is active bleeding or intracranial hematomas, is common. Hypofibrinogenemia with fibrinogen levels <100 mg/dL is treated with cryoprecipitate.
Intracranial bleeding is common after TBI and can worsen or be delayed. Hemostatic drugs may decrease the incidence or size of intracranial bleeds; however, a recent review concluded that there is no reliable evidence from randomized controlled trials that hemostatic drugs (aprotinin, tranexamic acid, aminocaproic acid, or recombined activated factor VIIa [rFVIIa]) reduce mortality or disability after TBI.61 Well-designed trials will be needed to assess the utility of these agents after TBI.
There is a wide variability in the reported incidence of early (within 7 days; 4%-25%) versus late (>1 week; 9%-42%) posttraumatic seizures (PTS) in untreated patients.62 The incidence of seizures following penetrating TBI is about 50% in patients followed for 15 years.62,63 Risk factors for seizures after TBI include penetrating injuries, cortical contusion, depressed skull fractures, intracranial hematomas (epidural, subdural, intracerebral), seizures within the first 24 hours of injury, GCS <9, and associated medical problems.63-65 Seizures early after TBI may be associated with conditions that can result in further secondary brain injury such as hypoxemia, hypercarbia, hypertension, increased ICP, increased cerebral metabolic rate, and excess release of toxic neurotransmitters. Antiepileptic drugs are associated with adverse side effects including skin rash, drug fever, elevated liver function tests, hematologic abnormalities, ataxia, and neurobehavioral side effects.
A large randomized, double-blind, placebo-controlled trial demonstrated that phenytoin prophylaxis results in a significant decrease in the incidence of early PTS (14.2%-3.6%) but no significant reduction in the incidence of late PTS, despite therapeutic phenytoin levels in most of these patients.63 The adverse drug effects during the first 2 weeks of treatment were not significantly different for placebo versus treated patients. The overall mortality rates were also not significantly different. Another smaller study also found no significant reduction in late PTS using phenytoin or phenobarbital prophylaxis.66 The majority of information is on clinically evident PTS. There is a paucity of data concerning nonconvulsive seizures after TBI67,68 and studies of the role of continuous electroencephalography monitoring post TBI and antiseizure therapy are needed.69,70
Currently, antiseizure prophylaxis with phenytoin is recommended for the prevention of early PTS, that is, within 7 days of the TBI. Routine prophylaxis later than 1 week following TBI is not recommended.62 Levetiracetam is a newer antiepileptic agent that has been evaluated for prophylaxis in TBI and appears to be effective.71,72 However, further studies are needed to establish the efficacy of levetiracetam as monotherapy.73 Phenytoin appears to be more cost-effective than levetiracetam.74 Levetiracetam may be reserved for patients with adverse reactions to phenytoin.
If late PTS occur, they should be managed with the standard approach used for new onset seizures.
TBI, as with other tissue injuries, is associated with complex inflammatory pathways involving pro- and anti-inflammatory cytokines, free radical formation, complement factors, adhesion molecules, and other pathways.75 Glucocorticoids have anti-inflammatory properties, can reduce free radical production, and have been shown to reduce ICP by reducing vasogenic edema associated with brain tumors76; however, there is no evidence that glucocorticoids reduce the cytotoxic edema associated with TBI or improve the clinical outcome.77,78 Trials using high-dose methylprednisolone,79 high-dose dexamethasone,80 the synthetic glucocorticoid triamcinolone,81 and the 21-aminosteroid tirilazad82 have not demonstrated an overall beneficial effect of steroids on outcome.78 There is level I evidence that high-dose methylprednisolone increases mortality after moderate to severe TBI, although the cause was not apparent.78,79 The negative results of corticosteroid trials may be related to their side effects and trials of more targeted anti-inflammatory agents are needed.75
Severe TBI sets into motion a cascade of injurious events including inflammatory, excitotoxic, edema forming and apoptotic processes7 that result in an imbalance between cerebral oxygen and nutrient supply and brain tissue metabolic demand. Intracranial hemodynamics is also dependent on alterations in systemic hemodynamics resulting in a very complex milieu.
Prevention of secondary injury via the early detection, treatment, and possible prevention of adverse intracranial pathophysiological events are the goals of bedside neuromonitoring. The brain, however, is a very delicate structure surrounded by blood vessels and encased within a bony vault and remains very difficult to safely and accurately interrogate at the bedside. Multimodality neuromonitoring aims to improve the reliability of information by simultaneously using two or more techniques to assess the state of the intracranial environment, the brain, and the response to therapeutic measures or changes in systemic hemodynamics.
Typical neuromonitors include measurement of ICP—the most commonly used neuromonitor in TBI, quantitative or qualitative CBF, jugular venous bulb oximetry [Math Processing Error], and brain tissue oxygenation [Math Processing Error]. Continuous EEG monitoring can be used in the detection and treatment of nonconvulsive seizures and status epilepticus.68 Other potential monitoring techniques have significant technical limitations (eg, cerebral oximetry via near infrared spectroscopy [NIRS])83 or are primarily research tools (eg, cerebral microdialysis).
The parameters to measure, the devices proposed to monitor them, the integration of this data, and its application to bedside management remain the subject of much clinical and laboratory research. Currently there are no clear data to support the use of a particular parameter, intervention or device.5,84-87 As such, the choice of monitor(s) depends on the technology available, the preferences and expertise of the staff, and individual patient considerations. The integration of information from multiple monitors in real-time using bioinformatics techniques to analyze data has been proposed88; however, improvements in the individual monitoring technologies will also be needed to improve clinical care. A brief description of the major bedside neuromonitoring modalities organized by parameter is provided below.
INTRACRANIAL PRESSURE MONITORING
Intracranial pressure (ICP) monitoring remains a cornerstone in neuromonitoring after severe TBI. The normal ICP in a supine adult ranges from 7 to 15 mm Hg.89 Elevated ICP or intracranial hypertension (IH) (ie, ICP >20-25 mm Hg) causes brain injury by ischemic mechanisms either by reducing cerebral perfusion or causing herniation of brain tissue, compressing the brain stem and leading to cardiopulmonary arrest. IH after TBI indicates severe brain injury and is a major predictor of mortality and neurological morbidity.90-93 IH occurs in 40% of patients after severe TBI.94 After TBI, comatose patients (GCS ≤8) with an abnormal CT scan have the highest risk for IH (53%-63%).92 Patients with a normal CT scan on admission have a lower incidence of IH (13%).5 After TBI, when increases in the mean ICP in 5 mm Hg increments were compared against outcome using stepwise ordinal logistic regression, a 20-mm Hg cutoff was found to optimally predict poor outcome (GOS) in the largest prospectively collected, observational study to date.95 Smaller, noncontrolled reports also suggest a range of 15 to 25 mm Hg.86,96,97
After TBI, persistent ICP >20 is associated with poor outcome and there are limited data—class II and III level evidence—that patients responding to ICP lowering treatments have a lower mortality and better outcome.5,97-100 Level II evidence supports ICP monitoring in salvageable patients after severe TBI that have a GCS ≤8 after resuscitation and have CT evidence of edema, herniation, contusions, hematomas, or compressed basal cisterns5 or that have a normal CT scan, but are at high risk of developing intracranial hypertension—that is, have two or more of the following on admission: age >40, unilateral or bilateral motor posturing, or hypotension (SBP <90 mm Hg).5 In patients with traumatic subarachnoid hemorrhage (SAH), ICP monitoring was the first indicator of evolving lesions in 20% of the severe TBI group, with 80% of these having operative intervention.101 Patients presenting with diffuse axonal injury after TBI, without associated mass lesions, are less likely to develop ICP elevation and may not need ICP monitoring.102
There is no level I evidence, that is, randomized prospective controlled clinical trial, that treatment based on ICP monitoring improves outcome after TBI.5 The results of a recently published Washington University-sponsored multicenter, controlled trial of patients aged ≥13 with severe TBI randomized to treatment based upon direct intraparenchymal ICP monitoring (target ICP ≤20 mm Hg) versus protocolized care based on imaging and clinical examination demonstrated no significant between-group difference in survival, impaired consciousness, functional exam at 3 and 6 months post-TBI, or ICU length of stay. The use of hyperosmolar agents and hyperventilation was significantly higher in the imaging and clinical examination group. Thus, for patients with severe TBI, it appears that pressure-targeted ICP monitoring is not superior to care based on neurological exam and serial CT imaging.103
ICP cannot be reliably predicted by physical exam or CT scan alone.5 Treating for presumed intracranial hypertension without actual monitoring of ICP may lead to the inappropriate application of hyperventilation, hyperosmolar therapy,104 or sedation (barbiturates)105
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


