Relationship between globin chains in haemoglobin
The haemoglobin molecule changes configuration during oxygen uptake and release. Deoxyhaemoglobin is in the taut (T) configuration, while the binding of oxygen produces a more relaxed (R) configuration by breaking salt links in the α1β2 and α2β1 interactions. The first oxygen molecule binds relatively weakly to haemoglobin, since more salt links must be broken (and therefore more energy is required) compared with the subsequent two oxygen molecules, and less energy still is required for the binding of the fourth oxygen molecule. This sequential increase in oxygen affinity explains the sigmoid shape of the oxyhaemoglobin dissociation curve of haemoglobin (Figure 12.2). Myoglobin, being a single-chain molecule, has a hyperbolic-shaped oxyhaemoglobin dissociation curve.
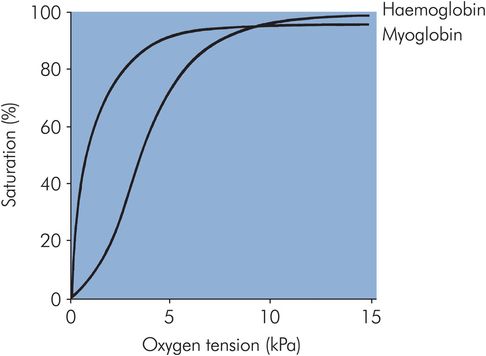
Oxyhaemoglobin dissociation curve
Haemoglobin is a good example of an allosteric molecule in that its oxygen binding properties are regulated by the binding of other substances at sites remote from the oxygen binding sites. These regulator substances are CO2, H+ and 2,3-diphosphoglycerate (2,3-DPG).
Effect of CO2, H+
CO2 binds to the terminal amino groups of haemoglobin as bicarbonate, which is formed in the red cell by the action of carbonic anhydrase:
This lowers the oxygen affinity of haemoglobin, shifting the oxyhaemoglobin dissociation curve to the right. Much of the hydrogen ion generated by this reaction is taken up by deoxyhaemoglobin, which has a higher affinity for H+ than oxyhaemoglobin:
Hence, under acid conditions the equilibrium between deoxy- and oxyhaemoglobin shifts in favour of deoxyhaemoglobin – this is called the ‘Bohr effect’. Acidic conditions, therefore, reduce the oxygen affinity of haemoglobin and shift the oxyhaemoglobin dissociation curve to the right. In the highly oxygenated environment of the alveolar capillaries of the lungs the above reactions are reversed.
Effect of 2,3-DPG
Metabolism in the erythrocyte is mainly via anaerobic glycolysis, following the Embden–Meyerhof pathway. 2,3-Diphosphoglycerate (2,3-DPG) is a product of glycolysis binding to haemoglobin in the ratio of one molecule of 2,3-DPG per tetramer.
2,3-DPG reduces the oxygen affinity of haemoglobin 26-fold (oxyhaemoglobin dissociation curve moves to the right). In the absence of 2,3-DPG, haemoglobin would unload little oxygen in the capillaries.
Part of the ‘storage lesion’ of blood for transfusion is a fall in 2,3-DPG levels to about 30% of normal after 3 weeks storage of whole blood in CPDA medium (citrate-phosphate-dextrose-adenine). This is improved with storage of plasma-reduced blood in SAGM (saline-adenine-glucose-mannitol). The clinical significance of the low 2,3-DPG is only likely to be of importance in recipients with severe anaemia or cardiac ischaemia. 2,3-DPG levels are restored to at least 50% of normal in 24 hours and 95% of normal at 72 hours after transfusion.
An increase in body temperature is capable of shifting the oxyhaemoglobin dissociation curve to the right and is an appropriate response to exercise. The converse is also true, but in hypothermic individuals the oxygen requirement also falls.
Genetic control of haemoglobin and haemoglobinopathies
The genes for the α-globins are located on the short arm of chromosome 16, while the β-globin gene cluster is located on the short arm of chromosome 11. Alteration of the coding regions (exons) of the globin genes is likely to alter the amino acid sequence of the corresponding globin chain, and the end result is a haemoglobinopathy (e.g. sickle cell disease). On the other hand, if there is lack of expression of a globin chain as a result of deletion or mutation at critical control regions (e.g. RNA splice sites) then a thalassaemia results. Although sickle cell and thalassaemia are the two most common anomalies of haemoglobin, there are other abnormal forms.
Sickle cell disease
Pathophysiology
Sickle cell disease is a result of a single DNA base change (adenine to thymine) which results in the substitution of valine for glutamic acid at position 6 of the β-globin chain.
Sickle haemoglobin (HbS) polymerises into microfibrils (crystals) in the deoxygenated state. It is thought that the formation of parallel HbS microfibrils causes red cell membrane damage, which results in the classical sickle cell deformity. The effect of this is to shorten the survival of sickle red cells (to 5–15 days in homozygous sickle cell disease), causing haemolytic anaemia. In addition, the deformed red cells are more rigid and less capable of passing through the microcirculation. The result is obstruction, clinically manifest as ‘crises’, e.g. bone pain, pulmonary syndrome and stroke.
The rate of polymerisation of HbS is related to the 50th power of its concentration. Polymerisation is also affected by the presence of other haemoglobin types, which may inhibit this process to varying degrees. Haemoglobin C and D are not sickling haemoglobins, but inhibit the sickling of HbS much less than HbA and HbF. Hence, persons with double heterozygosity for HbS and HbC or HbD have a sickling disorder, whereas heterozygotes for HbS do not.
Sickle cell disease exists widely throughout Africa and in parts of Asia, the Arabian peninsula and southern Europe. The high prevalence of this debilitating disease is a result of balanced polymorphism driven by the relative resistance of heterozygotes to malaria. The majority of sickle cell disease seen in the UK is found in African-Caribbean populations in large cities, where up to 10% of individuals carry the βS gene. In Africa this may be as high as 45%.
Clinical features: sickle trait
Sufferers of sickle trait have normal growth, development, exercise tolerance and life expectancy. There are rare reports of high-altitude splenic infarction, though it must be stressed that most persons with sickle trait tolerate high altitude with impunity.
Clinical features: sickle cell disease
The clinical features of sickle cell disease can be summarised as follows:
Anaemia – universal. Most patients have a haemoglobin level between 5 and 10 g dL–1. It is generally well tolerated and does not require therapy.
Vaso-occlusive (painful) crises – the most common manifestation that brings the patient into hospital contact. There is sudden onset of severe pain, usually in bones and joints, which reflects bone marrow ischaemia due to sickling in the bone marrow sinusoids. It may be precipitated by infection or exposure to cold, although often no precipitant can be identified. Abdominal crises may present as an acute abdomen and can be difficult to distinguish from biliary colic. The absence of bowel sounds, however, should alert the physician to a disorder requiring surgical intervention.
Acute chest syndrome – features include pleuritic chest pain, fever, tachypnoea, pulmonary infiltrates and leucocytosis. It is difficult to distinguish infection, infarction due to sickling, and pulmonary embolism. Hypoxia is common and ventilatory support is sometimes needed.
Stroke – perhaps the most important complication, with the majority occurring in children. Infarction is the usual pathology.
In children – aplastic crises (parvovirus), splenic sequestration (major cause of death in children under 2 years old), dactylitis, delayed growth and development.
Long-term complications in adults – include cholelithiasis, sickle retinopathy, leg ulcers (more common in the tropics), renal impairment and chronic bone damage from recurrent crises.
Diagnosis
Some other haemoglobin types may mimic sickle cell disease. To obtain an exact diagnosis, haemoglobin electrophoresis should be performed.
Medical management
Ideally all patients should be managed in centres with experience in managing haemoglobinopathies. Management of specific problems is as follows:
Vaso-occlusive crises – adequate and prompt pain relief is essential. Opioids are often required. Adequate hydration is essential. Broad-spectrum antibiotics should be given if infection is suspected. Hypoxia is the only proven indication for oxygen therapy.
Pregnancy – transfusion therapy during pregnancy reduces the incidence of painful crises but has not been shown to alter pregnancy outcome.
Prophylaxis – penicillin V 250–500 mg bd (important in hyposplenic status). Vaccination against pneumococcus, Haemophilus influenzae and meningococcus is recommended.
Perioperative management
Consult with haematologist preoperatively, to confirm diagnosis and possible antibodies due to previous transfusion.
Consider transfusion preoperatively to reduce the HbS level to < 30% and correct anaemia.
Special attention to be paid to hydration, oxygenation, hypothermia, acidosis preoperatively and postoperatively, as well as during surgery.
Increased infection risk requires antibiotic prophylaxis.
Monitor the use of tourniquets carefully and ensure effective exsanguination before inflation.
Thalassaemias
The thalassaemias are genetic disorders of globin chain synthesis (α or β). The genetic lesion results in failure of expression of the globin chain gene involved.
In the α-thalassaemias gene deletion is the usual mechanism, and in the β-thalassaemias gene mutation, resulting in abnormal processing, occurs most often. Heterozygotes are said to have thalassaemia trait; they are asymptomatic but may have characteristic blood count abnormalities.
Alpha-thalassaemia
In α-thalassaemias there is reduced or absent α-chain synthesis. It is critical in the understanding of these disorders to recall that there are two α-chain genes on each chromosome and therefore four genes in any diploid cell. Either one gene is deleted on the same chromosome (α+) or both are (α0). The homozygous condition in which no α chains are produced (—/— or α0α0) is not compatible with life, the fetus being stillborn at 28–40 weeks or surviving a few hours after birth. It is termed the Hb Barts–Hydrops syndrome.
Beta-thalassaemia
In β-thalassaemia there is reduced or absent β-chain synthesis. Each diploid cell has two β-chain genes. Mutation is the usual abnormality at the DNA level, which results in transcriptional dysfunction, RNA processing error or non-functional mRNA. This can result in either reduced β-chain production from that gene (β+) or absent production (β0). Each racial group affected has its own repertoire of mutations. Affected populations originate from the Mediterranean, Indian subcontinent or Southeast Asia.
Homozygous β-thalassaemia – thalassaemia major (Cooley’s anaemia)
There is absent or greatly reduced β-chain synthesis. This becomes apparent with the natural fall in fetal haemoglobin (α2γ2) levels due to the switch from γ– to β-chain production. The infant presents with anaemia during the first 6 months of life. If transfusion therapy is not instituted then the infant may demonstrate the typical thalassaemic facies (frontal bossing, maxillary hyperplasia). With transfusion therapy there is normal development for the first decade, but iron overload then becomes clinically apparent in the absence of chelation therapy (desferrioxamine) and death occurs in the second or third decade. Chelation is usually started in infancy, and with good compliance patients can expect to live well into their 30s and perhaps longer. In a select group of children bone marrow transplantation is an option, but not without risk of transplant-related death.
Beta-thalassaemia trait
This is asymptomatic with a thalassaemic blood picture. The HbA2 (α2δ2) level is elevated due to a relative reduction in β-chain synthesis with normal δ-chain synthesis. Iron deficiency can reduce HbA2 levels into the normal range, and therefore HbA2 should be measured for diagnostic purposes when the individual is iron-replete.
Transfusion medicine
Transfusion medicine involves the procurement, processing, testing and administration of blood and its components. It is also concerned with the prevention, investigation and treatment of transfusion-related complications.
ABO incompatibility remains an important cause of life-threatening transfusion reaction, and most often as a result of clerical error.
The blood group systems were recognised following a reaction between the recipient’s serum and donor red cells. Historically, this occurred in vitro and/or in vivo as a transfusion reaction or haemolytic disease of the newborn (HDN), such reactions being simple antibody–antigen reactions. If the particular antibody is able to fix complement (IgM, IgG), then lysis occurs. If this occurs in vivo, an immediate (intravascular) haemolytic transfusion reaction occurs.
ABO system
The ABO system is the most important blood antigen system, because of the presence of naturally occurring IgM antibodies to groups A and B in subjects lacking these antigens.
The IgM antibodies are active at 37 °C and readily cause immediate haemolytic transfusion reactions in ABO incompatibility. They are not present at birth, and are thought to arise after exposure to foreign antigens in infancy. Figure 12.3 gives the frequencies of the ABO groups in the UK. Among blood group A and AB individuals approximately one-quarter have a lower density of A antigen on the surface of the red cells, and such subjects are referred to as A2 and A2B (the rest are A1/A1B). Some A2 and A2B people have low levels of anti-A1 in their serum, though this is rarely of clinical significance as it is not active at 37 °C.
| Blood group | Naturally occurring antibodies (IgM) | UK (%) |
|---|---|---|
| O | Anti-A, anti-B | 47 |
| A | Anti-B | 42 |
| B | Anti-A | 8 |
| AB | None | 3 |
The majority (80%) of individuals can secrete ABO substances in their saliva and other bodily secretions. Persons who are group O and are secretors secrete a precursor substance (H) in the saliva, group A secretors secrete A and H, and so on.
Rhesus system
The rhesus sytem is the second most important system, not because of naturally occurring antibodies as in the ABO system, but because rhesus D (RhD) antibodies are readily formed when blood from a RhD-positive donor is infused into a RhD-negative recipient or when a RhD-negative mother bears a RhD-positive infant. Rhesus antibodies are IgG, and can therefore cross the placenta to cause haemolytic disease of the newborn (HDN).
Alongside the D antigen there are four other Rh antigens of importance – C, c, E, e – although a total of at least 50 Rh antigens have been described. The antigens C and c are allelic, as are E and e. There is no d antigen; the letter is used to indicate a lack of D antigen. D is the most important of the Rh antigens as it is more than 20 times more immunogenic than c, the next most important antigen. Because of the RhD testing of all donors and recipients, and immunoprophylaxis with anti-D during pregnancy and at delivery, the relative frequency of anti-D compared with other Rh antibodies has declined significantly in recent years. Red cell units are still labelled RhD-positive or negative, but a proportion will also have a Rh genotype label. The reason for this is to indicate a lack of certain antigens (c and e in the examples given); these units are, therefore, suitable for patients whose serum contains the corresponding antibodies. Numerous other red cell antigens also exist.
Group and screen
It is of critical importance that blood samples for compatibility testing are correctly identified. The ABO and RhD groups are established by using monoclonal typing reagents (anti-A, anti-B, anti-A+B and anti-D). In ABO grouping, the reverse group is also performed by mixing the patient’s serum with A1 and B red cells. Then an antibody screen is performed. The patient’s serum is tested against red cells that between them carry the antigens listed above. If there is a positive reaction in the antibody screen, then the antibody is identified by testing the patient’s serum against a panel of red cells. This is done either in the hospital blood bank or at the regional transfusion centre. Once an alloantibody is identified, appropriate red cell units, lacking the red cell antigen, are selected.
Red cell selection and cross-matching
If a red cell alloantibody is detected, then blood should be selected that is known to be negative for that antigen and is ABO- and Rh-compatible. A cross-match follows, where the patient’s serum is tested against red cells from the units to be transfused. If the cross-match is negative, then a compatibility label, giving patient details, is attached to the unit and the blood and a compatibility report issued.
Emergency situations
If the situation is life-threatening then group O blood is issued. If the patient is a premenopausal female, O RhD-negative must be issued. There is generally a shortage of O RhD-negative blood due to high demand for universal usage in emergency situations. Usually, there is sufficient time to perform ABO and RhD groups on the patient sample by rapid techniques and to do an immediate spin cross-match before issue. This may take 10–15 minutes. An antibody screen is then performed by the laboratory retrospectively. A label stating that standard pre-transfusion testing has not been performed is attached to the blood bag. In massive transfusion, where one blood volume has been given within 24 hours, ABO- and RhD-compatible blood can be issued without further serological testing, provided no alloantibodies were present by earlier testing.
Transfusion reactions
Immediate life-threatening reactions
Immediate haemolytic transfusion reactions
These are most often due to ABO incompatibility arising from clerical error. Less commonly, they may be due to antibodies against RhD, Duffy, Kidd and Kell antigens. The antibodies (IgM, IgG1 and IgG3) fix complement and this causes intravascular haemolysis. Features include pain at infusion site, chest and back pain, hypotension, DIC, haemoglobinuria. The mortality rate is 10%.
Delayed life-threatening reactions
Bacterial contamination
This is rare but is usually fatal if red cells are contaminated. One of the most frequent organisms responsible is Yersinia enterocolitica. There is rapid development of septic shock and collapse. The contaminated blood may be clotted or have a purple discoloration. Platelets can sometimes be contaminated with bacteria, most often Gram-positive organisms such as Staphylococcus epidermidis. This relates to storage of platelets at 22 °C.
Transfusion-related acute lung injury (TRALI)
This is rare. It is due to antileukocyte antibodies in donor plasma, and most often seen associated with fresh frozen plasma (FFP). Hypoxia develops during or in the hours following transfusion. There are associated bilateral lung infiltrates radiographically. This syndrome is easily confused with ARDS, and in the critical care setting TRALI may be overlooked as a cause of this clinical picture. Respiratory support is often required, and IV methylprednisolone should be given. In contrast with ARDS, recovery is usual within 48 hours. Recurrence is not a problem, as the antibodies existed in the donor plasma.
Congestive cardiac failure (usually LVF)
This is a common adverse effect of transfusion in the elderly, and can be late in onset. Patients with ischaemic heart disease are most at risk. It can be avoided by prophylactic diuretics and transfusion of only two units a day by slow infusion – one unit in 3 hours.
Non-life-threatening reactions
Febrile non-haemolytic transfusion reactions (FNHTR)
This is the most common type of transfusion reaction. It is due to recipient antileukocyte antibodies. These may be acquired following pregnancy or after transfusion of cellular blood components. The patient may experience a rigor, but temperature may rise without one. There are no signs or symptoms to suggest a haemolytic reaction, but severe FNHTRs may be associated with moderate hypotension, nausea, vomiting, cyanosis and collapse. If FNHTRs have already been experienced, further reactions can be prevented by paracetamol before transfusion. Leucodepletion of blood components may be indicated if reactions persist despite this measure. Primary prevention of FNHTR by leucodepletion is appropriate in some situations, e.g. aplastic anaemia, renal transplant candidates, transfusion-dependent patients.
Delayed haemolytic transfusion reactions
These reactions occur in patients who have been previously sensitised by transfusion or pregnancy. Red cell antibody titres in such patients may fall to undetectable levels over time, and subsequent re-exposure to the corresponding antigen in a later transfusion can result in a secondary immune response and haemolysis of the transfused cells. IgG antibodies are generally responsible, and haemolysis is generally extravascular, though occasionally it can be intravascular.
Clinical features include fever and a fall in haemoglobin level associated with jaundice between 4 and 14 days after transfusion. If the haemolysis is intravascular, then there may be haemoglobinuria. The clinical severity is related to the volume of incompatible blood transfused. Diagnosis requires a positive direct antiglobulin test and the demonstration of an alloantibody either in the serum or in a red cell eluate, which is antibody eluted from the red blood cells.
Use of blood components
Guidelines are available for the use of platelets, fresh frozen plasma (FFP) and cryoprecipitate. Figures 12.4, 12.5 and 12.6 give indications for the use of these components.
| Bone marrow failure – if this is reversible (e.g. after chemotherapy), prophylaxis to maintain platelets > 10 × 109 L–1 is appropriate. In chronic bone marrow failure platelets are generally given if the patient is haemorrhagic |
| Platelet function disorders – very occasionally required prior to surgery |
| Massive blood transfusion – clinically significant dilutional thrombocytopenia occurs after the transfusion of about 1.5 blood volumes. The platelet count should be maintained > 50 × 109 L–1 |
| Cardiopulmonary bypass surgery – platelet functional abnormalities and thrombocytopenia are common in this situation. Platelet transfusion should be reserved for patients with non-surgical bleeding. Prophylaxis is not indicated |
| Disseminated intravascular coagulation (DIC) – in acute DIC with haemorrhage and thrombocytopenia. Fibrin degradation products (FDPs) impair platelet function. In the absence of bleeding and in chronic DIC platelet transfusion is not indicated |
| Prior to surgery and invasive procedures – platelet count should be raised to 50 × 109 L–1. For operations on critical sites (e.g. brain and eye) the platelet count should be raised to 100 × 109 L–1 |
Note that a ‘standard dose’ of platelets in an adult may be considered as 4 units m–2.
| Replacement of single coagulation factor deficiencies where a specific concentrate is not available |
| Immediate reversal of warfarin effect – but in life-threatening bleeding due to warfarin prothrombinase complex concentrates (PCC = intermediate purity factor IX) and factor VII concentrates are indicated, together with vitamin K (5 mg IV) |
| DIC if there is haemorrhage and coagulation abnormality – if there is no haemorrhage or the condition is chronic, FFP is not indicated |
| Thrombotic thrombocytopenic purpura – a rare disorder which is treated with plasmapheresis using cryoprecipitate-poor FFP |
| Massive transfusion if there are abnormal coagulation tests (PT and/or APTT ratio ≥ 1.5) and fibrinogen > 1.5 g L−1 (if < 1.5 g L−1 cryoprecipitate indicated). Coagulation tests need to be repeated frequently to assess the need for further components |
| Liver disease – if there is bleeding, or prior to surgery/procedures if the PT ratio is prolonged ≥ 1.5 |
| Cardiopulmonary bypass – if there is non-surgical bleeding and a coagulation abnormality with normal platelet count and function |
Note that a ‘standard’ dose of FFP in an adult may be considered as 4 units.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


