Key Clinical Questions
Why do patients have acute flares of crystal arthropathies?
How is gout distinguished from other monoarthropathies?
What are the appropriate therapies for acute and chronic gout and pseudogout?
What are the pathophysiologic features of osteoarthritis (OA), and how do these relate to clinical manifestations?
How is OA differentiated from other types of arthritis?
What are the nonpharmacologic, pharmacologic, and surgical treatment options for OA?
Introduction
Gout, pseudogout, and osteoarthritis make up perhaps the largest portion of the rheumatic diseases that affect primarily joints. Although these three disease entities are quite different, they share a number of features in common: all tend to be diseases seen at older ages; all three are not uncommonly seen in overlap with each other; and all three are characterized primarily by inflammatory and/or mechanical abnormalities, rather than autoimmune ones. Whereas gout and pseudogout constitute diseases of abnormal crystal formation and its resultant responses, osteoarthritis is primarily a disease of cartilage loss and autodestruction. In the following sections, we discuss these three important diseases, their pathogenesis, and management.
Gout
Gout currently affects more than 3 million Americans, usually presenting with severe acute episodic arthritis, which may evolve into chronic destructive tophaceous disease. It is more common in men than women, and more common in African-Americans than whites. The prevalence of gout rises with age, from 17 per 1000 among individuals 45 to 64 years old, to as high as 41 per 1000 in those 75 years and older. When hospitalized for other conditions, elderly patients with gout are more likely to have their hospital discharge delayed than those without gout. The annual incidence of gout rose from 45 per 100,000 in 1977–1978 to 64 per 100,000 in 1995–1996, and is estimated to have continued to rise over the last decade. Overall, the prevalence of gout has nearly quadrupled over the past half-century. Despite being the most common inflammatory arthropathy, gout is frequently misdiagnosed and mistreated.
The most important risk factor for gout is hyperuricemia, or an excess of serum uric acid, the end product of purine metabolism. Serum concentrations are determined by the balance between urate production and elimination. Hyperuricemia may be caused by either overproduction or underexcretion of urate, or a combination of both. Consumption of meat or seafood promotes hyperuricemia and gout, a phenomenon related to the high purine content of these foods. Alcohol has been known to predispose to gout since the days of Hippocrates. Alcohol consumption increases urate production through multiple mechanisms, including generation and turnover of ATP, diuresis and dehydration, production of lactic and ketoacids (which block renal urate excretion), and the consumption of purines in alcoholic beverages. Beer and ale ingestion are most strongly correlated with the presence of hyperuricemia and gout (presumably because of their higher purine content), while hard liquor increases serum urate and gout risk to an intermediate degree. Moderate wine consumption has a lesser effect on serum urate and the risk of gout, possibly because of other compounds present in wine. (Historically, the high prevalence of gout in affluent drinkers of wine and port may be related to the illicit use of lead acetate by wine merchants as a preservative and sweetener; chronic lead poisoning causes tubulointerstitial kidney disease that promotes hyperuricemia.)
|
More than one hundred years ago Osler suggested that sugar intake might increase the risk of gout. Recent studies show that Osler was very likely correct. The prevalence of gout has increased over the last 50 years, concomitantly with obesity and diabetes, and the introduction and rising consumption of high-fructose corn syrup. Human metabolism and degradation of fructose generates uric acid to a greater degree than seen with other sugars. In addition, fructose may have other hyperuricemia-inducing effects by virtue of its ability to modulate urate transport in the kidney. Milk consumption appears to be protective against gout, perhaps because of the uricosuric effect of milk proteins such as casein.
Uric acid is eliminated from the body by both gastrointestinal and renal routes. Approximately one-third of urate elimination occurs through the gastrointestinal system, in saliva, gastric juices, pancreatic secretions, and direct loss from the bowel. The remaining two-thirds of urate excretion is renal, via glomerular filtration and a complex balance of tubular secretion and reabsorption. Tubular handling of urate is carried out via several organic anion transporters (OATs), including URAT1. A number of agents that stimulate renal urate excretion act by inhibiting URAT1 (Table 255-1).
| Urate-Increasing Agents | Urate-Decreasing Agents |
|---|---|
|
|
Hyperuricemia may be either primary or secondary. Primary hyperuricemia accounts for the vast majority of gout patients. About 90% of these patients are primary underexcreters, with genetic molecular defects in renal urate secretion. Most of these patients have otherwise normal renal function. Primary overproducers account for the remaining 10% of primary hyperuricemia. A few of these patients have rare neurobehavioral syndromes, such as complete hypoxanthine phosphoribosyltransferase (HPRT) deficiency (Lesch-Nyhan syndrome) or partial HPRT deficiency (Seegmiller-Kelley syndrome), that result in increased biosynthesis of purines and gout.
Secondary gout occurs when preexisting conditions result in decreased uric acid excretion or increased purine turnover. Acute or chronic renal insufficiency may impair urate excretion. Diseases of cell turnover, such as malignancies and hemolytic anemia, result in abnormally increased urate production. Gout can manifest in the early teens due to a group of autosomal dominant diseases, which include familial juvenile hyperuricemic nephropathy (FJHN) and medullary cystic kidney type II disease. These diseases feature a mutant gene that interrupts the tertiary structure of uromodulin, also known as Tamm-Horsfall protein. The clinical presentation of these diseases not only includes early-onset gout, but also progressive renal failure and polyuria.
The clinical evolution of gout generally occurs in four stages. In asymptomatic hyperuricemia, the risk of an acute gout attack increases as the level of uric acid rises past its solubility point (> 6.8 mg/dL). Once solubility is exceeded, monosodium urate crystals may precipitate in joint spaces and lead to acute gouty arthritis. During this stage, the innate immune system initiates a cascade of events, including activation of complement and resident joint tissue macrophages, and recruitment of neutrophils from the bloodstream. Recent studies have implicated the inflammasome, an intracellular assembly of macrophages and monocytes that activates interleukin-1β (IL-1β), in the inflammatory response to uric acid crystals. IL-1β stimulates the production of other inflammatory mediators, including tumor necrosis factor (TNF)-α, IL-6, IL-8, and prostaglandin E2. This results in severe local inflammation as well as systemic response to cytokine release, such as fever and elevated acute phase reactants. These attacks are exquisitely painful but typically self-limited, even without therapy, perhaps because of immune autoregulation.
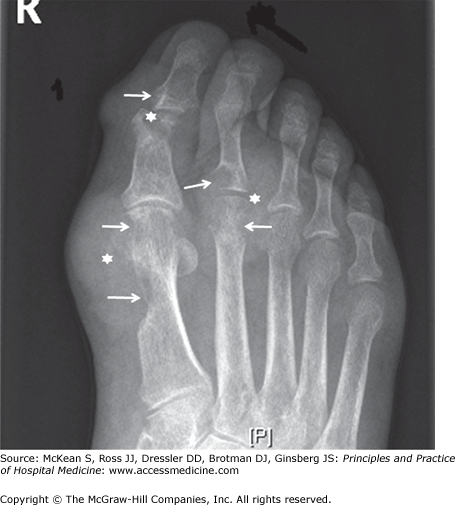
The asymptomatic interval between acute gouty attacks is known as intercritical gout. Over time attacks tend to come more frequently, the intercritical period dwindles, and chronic tophaceous gout may develop. Tophi are aggregates of urate crystals, typically accompanied by a low-level chronic inflammatory state. They are actually complex structures, consisting of a mix of monosodium urate crystals and cellular debris surrounded by activated macrophages. Although tophi are most obvious when they occur in the periarticular soft tissues, they may also develop in cartilage and bone. Radiographic images of tophi in periarticular bone characteristically reveal the pathognomonic finding of punched-out erosions with sclerotic margins and overhanging edges (Figure 255-1).
The gold standard for diagnosing all forms of gout is needle aspiration of acutely or chronically inflamed joints or tophi, followed by polarized light microscopy to identify negatively birefringent, needle-shaped crystals (uric acid crystals). The presence of such crystals, particularly when seen intracellularly within infiltrating neutrophils, confirms the diagnosis of an acute attack and helps to distinguish gout from septic arthritis, pseudogout, and other causes of inflammatory joint disease. (Extracellular crystals alone are not diagnostic of an acute gouty attack. In patients with established gout, extracellular crystals may persist even after inflammation has resolved, residing as “innocent bystanders” in the setting of other acute joint pathologies.) The sensitivity of synovial fluid analysis for demonstrating negatively birefringent crystals in patients with acute gouty arthritis is at least 85%, with a specificity approaching 100%. In contrast, the specificity of a clinical diagnosis of gout is significantly lower, and septic arthritis and pseudogout may be misdiagnosed as gout. Therefore, in almost all circumstances, diagnostic arthrocentesis should be performed.
As acute gout may coexist with other joint pathology, a wider evaluation is warranted, even in the presence of intracellular, negatively birefringent crystals. In addition to crystal analysis, synovial fluid should always be sent for cell count with differential, Gram stain, and culture. Grossly, synovial fluid is typically straw colored and varies from translucent to opaque. Synovial fluid cell counts in gout usually range from 2000 to 100,000 per mm3, with greater than 50% neutrophils (usually approaching 90%). Serum uric acid can also be obtained during an acute attack. A high serum urate supports the diagnosis of gout. However, levels may be paradoxically normal or low during an acute attack due to an increase in renal excretion, rising again two weeks after the attack. Erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) are often high, but this finding is very nonspecific. Urinary urate collections should not be obtained during gout attacks; they are neither useful nor reliable in the acute period.
Radiographs in the acute setting are not generally helpful, but radiographic evidence of erosions indicates chronic disease. Ultrasound is gaining popularity in the evaluation of musculoskeletal disease, including crystal-induced arthropathy. Musculoskeletal ultrasound (MSUS) has the capacity to visualize intraarticular crystal deposits, with a characteristic hyperechoic enhancement of the outer surface of the hyaline cartilage, known as the double contour sign. MSUS may prove to be an alternative method for the diagnosis of the crystal arthropathies, but limitations include the inability to distinguish the presence or absence of infection.
The differential diagnosis for acute gout includes not only conditions generally associated with acute monoarthritis, such as pseudogout and septic arthritis, but also conditions leading to oligoarthritis and polyarthritis, such as reactive arthritis, psoriatic arthritis, and even rheumatoid arthritis. Ideally, a crystal diagnosis of gout should be the goal; if this is not possible, the diagnosis of acute gouty arthropathy should be made by a combination of historical and clinical criteria. A thorough history will help distinguish an acute gout attack from other causes of acute arthritis. Some of the previously proposed clinical, radiographic, and laboratory criteria include (1) a history of one or more episodes of monoarticular arthritis, followed by intercritical periods completely free of symptoms (may not be applicable in the hospital setting); (2) maximum inflammation within 24 hours of onset of the attack; (3) rapid resolution after initiation of colchicine or a nonsteroidal anti-inflammatory drug (NSAID); (4) unilateral first metatarsophalangeal joint attack (podagra), especially if it is the first event; (5) hyperuricemia; and (6) subcortical bone cysts on plain radiograph.
Acute attacks most often affect the first metatarsophalangeal joint (up to 50% of first attacks). The tarsal joints, ankles, knees, elbows, and interphalangeal (IP) joints are also commonly affected. In elderly patients with osteoarthritis (OA), Heberden nodes or Bouchard nodes are potential targets for inflammation, and red, swollen proximal and distal IP joints may be the first manifestation of gout. Episodes of acute gouty arthritis frequently begin suddenly at night or in the early morning with dramatic pain and swelling. The joint rapidly becomes warm, red, and tender, often mimicking cellulitis. Without treatment, most acute attacks resolve in 3 to 10 days (range, 2 days to 2 weeks). Most patients, at least initially, do not have residual symptoms once an attack is complete (Table 255-2).
|
While acute attacks of gout can usually be managed in the outpatient setting, patients may need to be admitted to the hospital to facilitate workup or to rapidly and definitively rule out the possibility of infection. Patients with severe disability from their acute attack and inadequate home support may need to be admitted until they are able to ambulate or otherwise function. More often, patients at risk may develop acute gouty attacks while hospitalized for other problems. In the hospital setting, attacks may be precipitated by illness-related issues (eg, metabolic acidosis), diuretics or other medications, or disruptions in volume status or renal function.
Fluctuations in serum urate levels are associated with acute gout attacks. For this reason, patients with chronic gout should have their urate-lowering medications or other prophylaxis continued during the hospital stay, unless otherwise contraindicated (eg, in the setting of acute kidney injury). For example, stopping allopurinol in hospitalized patients may precipitate acute gout and increasing allopurinol doses during an acute attack could exacerbate the existing flare and prolong the clinical course.
Colchicine is effective in the acute setting, and, if initiated sufficiently early, may abrogate an acute attack. It is typically less effective after the attack is established. The traditional dosing of colchicine for acute attacks (0.6 mg by mouth every one to two hours) until onset of relief; nausea, vomiting, or diarrhea; or a maximum of 10 doses frequently incurred undesirable side effects. Recent studies suggest that a more limited regimen—colchicine 1.2 mg, followed by 0.6 mg one hour later—is equally effective as the old regimen, with much less toxicity. It has now been adopted as standard of care. Patients with renal insufficiency may receive this regimen, but should subsequently temporarily discontinue any use of low-dose daily colchicine for future prophylaxis; patients without renal disease may continue their prophylaxis unabated. Intravenous colchicine curtails gastrointestinal side effects but carried a much higher risk of toxicity and even death; its use has been discontinued by the U.S. Food and Drug Administration.
|
For many patients, NSAIDs
Related posts:
 Strategies for Cost-Effective Care
Strategies for Cost-Effective Care
 Building, Growing, and Managing a Hospitalist Practice
Building, Growing, and Managing a Hospitalist Practice
 Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
 Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


