(1)
Pediatric GI, Massachusetts General Hospital, 175 Cambridge ST, Room 575, Boston, MA 02114, USA
(2)
GI Unit, Massachusetts General Hospital, GRJ 719, 55 Fruit St, Boston, MA 02114, USA
Keywords
Visceral painIrritable Bowel syndromeSensitizationBacterial floraChronic painIntroduction
Nocioception is the detection of noxious stimuli [1] and acute nociceptive pain is produced when a noxious stimulus of enough intensity activates receptive pathways by damaging or threatening to cause tissue damage [2]. This mechanism is protective and helps prevent injury or further lesion by generating a reflex withdrawal and thus removal an offending stimulus. Not only is there a sub-conscious reflex elicited, but often the development of complex behavior or strategy in response to the unpleasant sensation with the principal goal of avoiding further damage and limiting injury. There are, however a variety of pain syndromes, some of which involve the viscera, where no significant gross tissue injury or structural disease is found even when using careful clinical methodology. Pain syndromes can also present despite the fact that inflammation or tissue damage has resolved. Such is often the case, for example, with functional gastrointestinal disorders and chronic abdominal pain. Functional chronic abdominal pain is formally defined by the ROME III Criteria as severe, usually generalized abdominal pain which is continuous and affects functioning and quality of life [3]. This is in contrast to functional dyspepsia (FD) and irritable bowel syndrome (IBS), which are intermittent. These abdominal pain syndromes such as FD and IBS are very common, so much so that in a study of consecutive outpatient visits to a university hospital, approximately 40 % of patients with a chief complaint that included abdominal symptoms were diagnosed as having a functional gastrointestinal disorder [4]. FD is a syndrome with symptoms centered in the upper abdominal region and include pain, heartburn, postprandial discomfort or bloating, and a heavy feeling in the stomach or fullness [5]. IBS is a syndrome that is characterized mostly by lower GI tract symptoms including abdominal pain or discomfort and altered bowel habits such as constipation and/or diarrhea, urgency and tenesmus [6]. The Rome III Diagnostic criterion for IBS is: Recurrent abdominal pain or discomfort at least 3 days/month in the last 3 months associated with two or more of the following: (1) Improvement with defecation. (2) Onset associated with a change in frequency of stool. (3) Onset associated with a change in form (appearance) of stool [7]. The Diagnostic criteria for FD, according to the Rome III criteria, must include one or more of the following: bothersome postprandial fullness, early satiation, epigastric pain, or epigastric burning and no evidence of structural disease (including at upper endoscopy) that is likely to explain the symptoms. This must be for the last 3 months with symptom onset and at least 6 months prior to diagnosis [8]. The different syndromes that comprise the functional gastrointestinal disorder spectrum are all made as diagnoses of exclusion, thus absence of an organic cause such as an ulcer, esophagitis, celiac disease, or cancer is important.
The cause and pathophysiology of FD is not completely defined, however several pathogenic factors have been proposed including motility abnormalities, visceral hypersensitivity, psychosocial factors, excessive gastric acid secretion, Helicobacter pylori, genetics, environment, diet, lifestyle, and post-infectious FD. It is likely that several factors may be involved even in the same individual. Many of these factors are also common to other functional gastrointestinal disorders and visceral pain syndromes [5].
Perception of pain caused by an innocuous peripheral stimulus, for example mechanical stimulation at a lower threshold than normal, in subsets of patients with IBS suggests abnormal processing of sensory information. This can occur by both the peripheral nervous system, and/or the central nervous system. A number of different potential mechanism have been proposed in IBS, including (1) a peripheral sensitization of sensory endings present in the gut wall; (2) increased flow of nociceptive information traveling through the sensory afferents at the level of the dorsal root ganglia or the nerve fibers of the exterior laminae of the spinal cord; (3) a reduced antinociceptive effect of descending inhibitory pathways acting in the spinal cord, and (4) a central amplification of afferent signals (anticipation and hypervigilance), possibly influenced by psychological factors as for example anxiety or depression [9]. We will review some of these, in particular peripheral and central sensitization, which appear to be key in the development of visceral pain.
Motility Abnormalities
The incidence and causality of motility abnormalities in FD has been investigated extensively and in different subsets of patients. A long list of abnormalities has been found including: impaired fundic accommodation, antral hypomotility, decreased antral distention, gastric dysrhythmias, and small bowel dysrhythmias [10], (Fig. 1.1). Some groups report a high prevalence of some or many of these specific abnormalities. In IBS, constipation or diarrhea can occur secondary to disordered motility from the small or large bowel (Fig. 1.2). Tack et al. found that 40–50 % of FD patients show impaired gastric accommodation, thought to cause early satiation [11]. The observations from different studies however have not been consistent and the strength of association of each of the motility disorders is not yet well defined. In a similar manner, delayed gastric emptying for solids has been found in approximately 40 % of FD patients [12], but the direct relationship between gastric emptying and dyspeptic symptoms is unclear [13]. Thus although motility disorders are common, their frequency and characteristics in each of the functional gastrointestinal pain syndromes vary and is still subject of debate. Additionally, it is unclear if motility abnormalities are a cause or an effect of the underlying pathogenic mechanism.
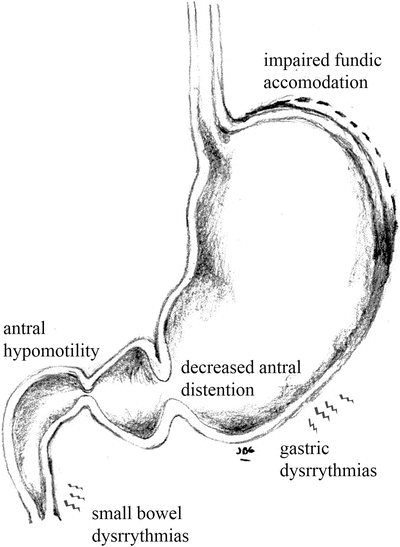
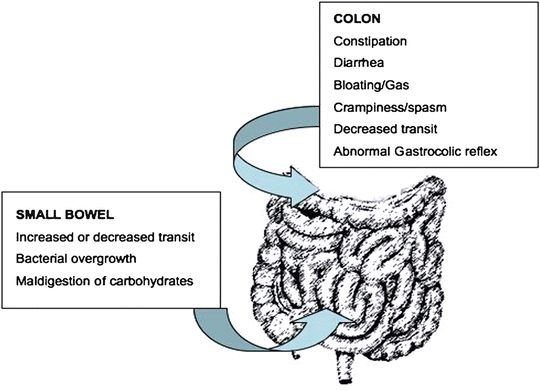
Fig. 1.1
Motor abnormalities in upper GI tract in FD
Fig. 1.2
Motor abnormalities and some associated conditions, in the lower GI tract in IBS
Sensitization
As a mechanism of added protection, sensitization is a physiological mechanism that enhances the nociceptive system [14]. Sensitization usually occurs immediately after exposure to an intense noxious event or after a repetitive damaging stimulus. When sensitization occurs, there is a reduction of the threshold needed for activation of the nociceptive response and in addition, an amplification of subsequent stimuli [15]. Sensitization is an adaptive process, which can occur in both normal and pathologic conditions and has the purpose of making the system hyper-alert to avoid ongoing damage where there is risk of further injury. It requires central nervous system synaptic, structural and chemical plasticity and although not necessarily permanent, some changes may be persistent [1]. In general, if the tissue injury or offending insult ceases, the state of heightened alertness returns over time to baseline and thus high-intensity stimuli are again required to initiate the response. In abnormal circumstances however, the state of heightened alertness becomes persistent despite an absence of ongoing tissue injury or of persistent nociceptive stimuli (Fig. 1.3).
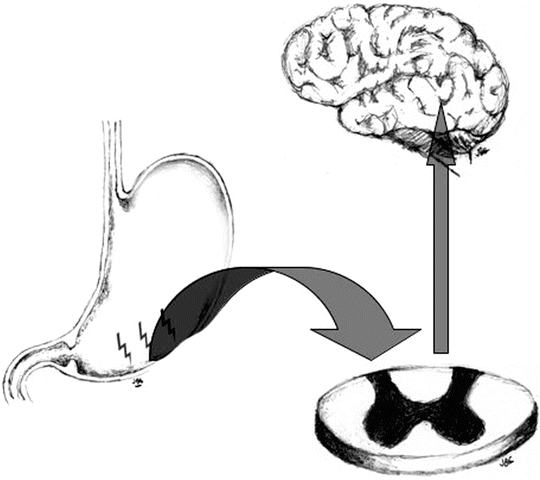
Fig. 1.3
Sensitization involves central nervous system synaptic, structural and chemical plasticity
Painful Syndromes that Involve Sensitization
Several painful syndromes including neuropathic pain [16], inflammatory pain [17, 18], migraine [19–21], and some types of headache [22], IBS [23, 24], fibromyalgia [25, 26], osteoarthritis [27], musculoskeletal disorders [28], generalized pain hypersensitivity [1, 29], temporomandibular joint disorders [30, 31], dental pain [32, 33], visceral pain hypersensitivity disorders [34–36], and postsurgical pain [29, 37–39] have been found to involve central sensitization. For example, visceral hypersensitivity thought to be determined by both central and peripheral mechanisms has been described in 20–90 % of patients with IBS [40]. Direct imaging of brain activity using functional magnetic resonance or positron emission tomography has demonstrated abnormal brain processing of peripheral sensory input [41]. In this scenario, the CNS malfunctions and the pain is no longer protective, on the contrary, it can be transformed to a new type of symptom often bothersome or worrisome to the patient and which may interfere with daily activity. In extreme cases, the pain or discomfort perceived may be severe. In these instances, the pain arises spontaneously or can be elicited by normally innocuous stimuli, a process referred to as allodynia. When the painful sensation is exaggerated and/or prolonged in response to noxious stimuli it is known as hyperalgesia, and when it spreads beyond the site of injury as secondary hyperalgesia (Fig. 1.4).
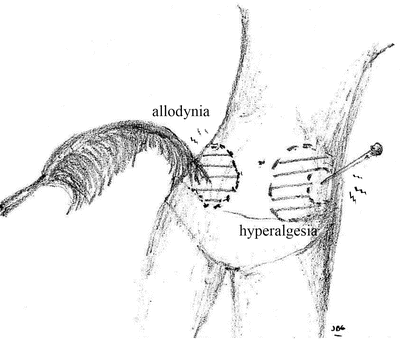
Fig. 1.4
Pain, which arises spontaneously or elicited by normally innocuous stimuli, is called allodynia. Hyperalgesia occurs when a painful sensation is exaggerated and/or prolonged
Pathophysiology of Peripheral and Central Sensitization
Abnormal pain sensitivity is due to peripheral receptor and CNS changes a process known as peripheral and central sensitization respectively. As mentioned above, sensitization is believed to be an important mechanism explaining many acute and chronic pain syndromes. Peripheral and central sensitization differs both on the mechanisms that are involved in the pathogenesis as well as the manifestations that elicited. Peripheral sensitization involves mechanisms that lower the threshold of activation and amplify the response of pain signaling through a modification of the peripheral nocioception receptors [42, 43]. These receptors are high-threshold primary sensory neurons, which can undergo a change when exposed to inflammatory mediators and/or damaged tissue. This process is generally limited to the area that is injured or inflamed [43] and pain hypersensitivity or primary hyperalgesia at inflamed sites generally requires ongoing pathology for its maintenance. In certain cases, however the hypersensitivity may be longer lasting, for example in altered heat sensitivity where peripheral sensitization appears to play a major role. This is opposed to mechanical sensitivity which is a major feature of central sensitization [1].
In IBS the contribution of peripheral factors to pain perception has been increasingly recognized as being very important, since a subset of patients can develop IBS following and apparently as a result from an acute episode of infective gastroenteritis (i.e., post-infectious IBS). There is also additional evidence for gut immune, neural, endocrine and microbiological (i.e., intestinal microbiota) abnormalities in large subsets of patients. These factors likely influence each other and play an important contributing role in pain transmission from the periphery to the brain via sensory nerve pathways [9].
Inflammation in various areas of the GI tract as part of different disease states has been associated with the development of visceral hypersensitivity and pain as well. These diseases include: reflux esophagitis, Helicobacter pylori gastritis, celiac disease, acute infectious gastroenteritis, and inflammatory bowel disease [40, 44]. Using animal models, a wide array of mediators released by inflammatory cells have been found to induce or mediate the peripheral sensitization of mucosal neuronal afferents [44] and to recruit nociceptors that were previously silent [44]. These mediators include: cytokines, prostanoids, amines, neuropetides, and neurotrophins. Similar mechanisms may also cause altered motor and not only sensory function in post-infectious IBS [45, 46]. Stimulated by chemical and/or mechanical stimuli, terminal receptors of visceral afferent nerves respond by activation of ion channels which induces sensory transduction [47]. These receptors are located in the nerve terminals in the gut mucosa, muscle, and serosa and transmit sensory information from the viscera to the central nervous system through the vagal and spinal afferent nerves.
Recent studies have found evidence that help explain peripheral sensitization through changes in the enteric nervous system and/or afferent pathways in several visceral pain syndromes including functional gastrointestinal disorders like IBS. For example, an increased expression of transient receptor potential vanilloid type-1 (TRPV-1) has been described in animal models of intestinal inflammation [48]. TRPV-1 is an ion channel receptor activated by heat (>43 °C), acid (pH <5.9), inflammatory mediators, and capsaicin [49]. Increased levels of TRPV-1 were also found in patients with idiopathic rectal hypersensitivity and fecal urgency [48]. A correlation between increased expression of TRPV-1 and rectal distension sensory and health thresholds has been described [50].
In the past few years, new and exciting information has become available, shedding light into the mechanisms of bidirectional brain-gut interactions in both health and disease. Evidence is compelling for enteric flora including both commensal and pathogenic organisms to exert an important role in the brain-gut interaction in both directions: brain-gut and gut-brain [51, 52]. The brain influence on the intestinal microbiota can be through direct or indirect mechanisms. Indirect ways to influence bacterial composition and function include motility, secretion, and intestinal permeability [53]. Direct pathways can occur via signaling molecules secreted into the gut lumen by various types of cells in the mucosa, lamina propria, and even myenteric and neuronal layers of the gut. [54] Conversely, the influence of bacteria upon the intestine and eventually the brain occurs via signaling molecules which exert their actions through specific receptors expressed on gut epithelial cells or enterochromaffin cells. In cases of increased intestinal permeability which can occur with inflammatory states such as invasive infectious diarrhea, bacteria can directly stimulate host cells in the lamina propria.
A key element in bidirectional brain-gut communication is the enterochromaffin cell, functioning as a transducer for the communication between the gut lumen and the nervous system. Afferent stimuli involving pain and immune-response modulation and even background emotions as well as other functions are transmitted from enterochromaffin cells via their direct innervation by vagal branches. Disruption or abnormalities in the bidirectional interactions between the bacteria and the brain contribute to several acute and chronic gastrointestinal disease states which include, for example IBS and other functional as well as inflammatory bowel disorders [55]
The bidirectionality of brain-gut information may help explain the results of a prospective 12-year study on a random population of 41,775 who were surveyed in Australia. The investigators found that among people free of functional gastrointestinal disease (FGID) at baseline, higher levels of anxiety was a significant independent predictor of developing new onset FGID 12 years later. Conversely, among people who did not have elevated levels of anxiety and depression at baseline, those with a FGID at baseline had significantly higher levels of anxiety and depression at follow-up. In IBS, higher levels of anxiety and depression at baseline were predictive of IBS at follow-up, while only depression was predictive of FD at follow-up. [56]
In Central Sensitization, the pain occurs due to CNS dysfunction, with both brain and spinal cord undergoing changes that lead to an alteration of how it responds to sensory inputs. It does not depend on the presence of peripheral noxious stimuli, as in the case of Peripheral Sensitization. The hypersensitivity response in non-inflamed tissue occurs due to a change in the sensory response, which is elicited by normal inputs. This leads to increased pain sensitivity, often long after the initiating cause has disappeared and when no peripheral pathology is present. Thus Central sensitization is an abnormal state of increased or amplified responsiveness to peripheral signaling caused by increases in membrane excitability, synaptic efficacy and/or reduced inhibition.
The mechanics of this process involve the recruitment of inputs, which do not normally activate nociceptive pathways, like for example: large, low-threshold mechanoreceptor myelinated fibers which go on to transmit fiber-mediated pain. In this manner, mechanical sensitivity becomes a major feature of central sensitization. Since this process requires a change in the neuronal properties of the CNS, the sensation of pain is no longer proportional nor does it correspond to the intensity, or duration of the different peripheral stimuli, as normally occurs in acute nociceptive pain.
In central sensitization, somatosensory neurons that are not fixed or hard-wired, but are instead highly malleable go on to sensitization by synaptic enhancement or increased efficiency [1]. When this process becomes established pain is elicited, which is erroneously perceived as a peripheral nociceptive event, since often there is no abnormal peripheral event occurring. Rather there is an abnormal response to both innocuous and/or noxious stimuli with a spread of tenderness or pain beyond lesion sites. Therefore, when tissue inflammation or disease has been ruled out and the diagnosis is deemed to be central sensitization, the treatment for this altered sensory condition is aimed at the CNS and not at peripheral nociceptive receptors or peripheral tissue.
A growing body of evidence has shown that this process is not restricted to neurons, as glial activation plays an important role in the modulation of neuronal functions and affects the spinal processing of nociceptive signaling. Indeed, glia are also involved in central sensitization and chronic pain facilitation. For example, glial activation in the spinal cord is considered an important component in the development and maintenance of allodynia and hyperalgesia in various models of chronic pain, including: neuropathic pain, pain associated with peripheral inflammation, and some forms of visceral hyperalgesia [57].
For central sensitization to be induced, an intense, repetitive, or sustained noxious stimulus is required recruiting many neural fibers. This will not occur after a single stimulus. Important tissue injury almost always induces central sensitization, so that for example surgical trauma will often result in the development of central sensitization, but peripheral tissue injury is not always necessary. Nocioceptor afferents from different parts of the body may be involved, for example: those innervating muscles [18, 58], joints [17], skin [59, 60] or internal organs [61, 36]. Experimentally, central sensitization can be elicited by nocioceptor activation of the skin, as with topical or intradermal capsaicin or with repeated heat stimuli [62–64] and in the gastrointestinal tract by exposure to low pH solutions [65, 66] or with mustard oil instillation into the colon in anesthetized rats [67]. Exposure of the lower esophagus to acid, for example, induces central sensitization leading to viscero-visceral (pain hypersensitivity in the upper esophagus) and viscero-somatic hypersensitivity (allodynia on the chest wall) that can be captured by esophageal evoked potentials, and is associated with increased temporal summation [68]. Thermal and mechanical pain hypersensitivity in the rectum after esophageal stimulation using acid and capsaicin infusions demonstrates how widespread the effects of central sensitization are in the gastrointestinal tract [68]. Indeed, 35–50 % of FD patients are said to be hypersensitive to gastric distension stimuli [69]. In a similar manner in IBS, several studies have found that balloon inflation in the sigmoid colon evokes increased pain perception compared with healthy controls [70]. In one series, rectal hypersensitivity could be detected in 95 % of patients with IBS [71].
Once afferents are stimulated with enough intensity/duration, there are two temporal phases that are triggered in central sensitization, each of which involves two specific mechanistic stages: An early transcription-independent, phosphorylation-dependent phase and a later transcription-dependent phase. The early phase is mediated by rapid changes in glutamate receptor and ion channel properties. The later is longer-lasting, requiring synthesis of new proteins [15] and is the mechanism responsible for central sensitization in several pathological syndromes and conditions [15]. To be established, central sensitization involves primarily, but not exclusively the N-methyl-d-Aspartate receptors (NMDAR) expressed in postsynaptic spinal cord neurons from the dorsal horn. Besides NMDAR, other receptors include: ionotropic amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA), Kainate (KA) receptors, and several metabotropic (G-protein coupled) glutamate receptor subtypes (mGluR). In the superficial laminae of the dorsal horn, AMPAR and NMDAR are present in virtually every synapse and these respond to the fast neurotransmitter glutamate, released by primary afferent neurons [72–74].
The activation of these receptors is very important at both initiating and also maintaining activity-dependent central sensitization. When using noncompetitive or competitive antagonists to experimentally block activation, the induced hyperexcitability of nociceptor conditioning is prevented [75]. Additionally, conditional deletion of NR1, the most common NMDA complexes in the dorsal horn, abolishes NMDA synaptic inputs and acute activity-dependent central sensitization [76], thus the NMDA receptor is both a trigger and an effector of central sensitization [1]. Stimulation of receptors: AMPAR and group I mGluRs [77–79] participate with NMDAR in the activation intracellular pathways, including the PLC/PKC pathway, the phosphatidylinositol-3-kinase (PI3 K) pathway and the mitogen-activated protein kinase pathway (MAPK) that involve the extracellular signal-regulated kinases (ERK1 and ERK2) and the cAMP response element binding protein (CREB). One way that ERK and CREB are activated is through an elevation in intracellular Ca2+ sufficient to drive a calmodulin-induced stimulation of adenylyl cyclases 1 and 8, whose cAMP production in turn activates PKA and subsequent cascade(s) [1]. Changes in the membrane receptors of the superficial lamina dorsal root neurons make previously Ca2+ impermeable to Ca2 + permeable AMPARs and significantly contribute to the source of the [Ca2+]i increase in pain, specifically that which is inflammation-induced.
Together with Glutamine, the neurotransmitters: Substance P (SP) [80, 81] and Calcitonin Gene-Related Peptide (CGRP) [82–84] are also important in the induction of central somatization. Additionally CGRP enhances brain-derived neurotrophic factor (BDNF) release into the spinal cord from nociceptive neurons in an activity-dependent manner. BDNF binds to its high-affinity tyrosine-kinase receptor trkB and enhances NMDA-mediated C-fiber–evoked responses and causes activation of several signaling pathways in spinothalamic track neurons, including ERK [85, 86] and PKC, thus also contributing to central sensitization [87, 88]. Other important mediators that activate the CNS pathways and mediate central sensitization include: the inflammatory kinin bradykinin [89, 90] produced in the spinal cord as a response to peripheral noxious stimuli [91], nitric oxide synthesized by either neuronal or inducible NO synthase in the dorsal spinal cord [92–94], and serotonin (5-HT) [95] primarily through the 5-HT3 receptor.
Serotonin (5-HT) is a very important neurotransmitter and paracrine-signaling molecule between the brain and the gut [96]. Approximately 90 % of the production of 5-HT in the human body is in gastrointestinal tract, mainly synthesized by enterochromaffin cells, a subtype of enteroendocrine cells [96]. The remaining 10 % is produced in the brain in which it acts as a neurotransmitter. Peripherally produced 5-HT does not cross the blood–brain barrier. In the GI tract 5-HT activates multiple receptors (5-HT1 to 5-H7) mostly expressed by different intrinsic primary afferent neurons within the enteric nervous system and also extrinsic afferent sensory nerves [96–98]. 5-HT modulates visceral sensation via both 5-HT3 receptor and non 5-HT3 receptor-dependent mechanisms on vagal or spinal afferents [99]. The activity of 5-HT is terminated by the serotonin reuptake transporter (SERT), which is expressed in the enterocyte. 5-HT has received a great deal of attention in gastrointestinal pain syndromes and several reports have described an important role for this molecule. Alosetron a 5-HT3 receptor antagonist, for example, inhibits spinal cord c-fos expression in response to noxious colorectal distension [100]. This observation suggests that 5-HT plays a role in the transmission of noxious information within the spinal cord. 5-HT3 receptor antagonists and 5-HT4 receptor agonists in human trials have also been found to decrease multiple IBS symptoms as well [6, 101].
As previously mentioned, there are multiple different ways to initiate and precipitate the series of reactions that contribute to the establishment central sensitization. These different processes elicited as response to nociceptor input, can (1) increase membrane excitability, (2) facilitate synaptic strength, or (3) decrease inhibitory influences in dorsal horn neurons [1]. Initial observations of central sensitization proved the plasticity of flexor motor neurons following peripheral nerve injury [102]. This led to finding similar changes in spinothalamic tract neurons [94, 103, 104], thalamus [105], spinal nucleus pars caudalis [106], anterior cingulate cortex [107], and amygdala [108, 109]. Thanks to modern imaging techniques such as functional magnetic resonance imaging, magnetoencephalography, and positron emission tomography, other brain structures involved in pain have demonstrated an increase in excitability and thus central sensitization. Amongst these are: the prefrontal cortex, superior colliculus, parabrachial nucleus, and periaqueductal gray area [110–116].
Central Sensitization in Pathological Settings
Phenotypic changes in myelinated fibers after inflammation and/or nerve injury can enable afferents to acquire the capacity to generate central sensitization. In this scenario, a normally protective mechanism may result in a pathological process. This most commonly occurs when inflammation or ongoing tissue injury persists. Additionally, there are abnormal situations where even in the absence of active peripheral pathology Central sensitization becomes autonomous and persistent and pain can be triggered not only by less intense inputs but also be maintained by different non-nociceptive stimuli. As an example: ongoing C-nerve fiber stimulation, even at levels that do not elicit central sensitization in basal conditions, is sufficient to maintain central sensitization once it has been induced for days [117]. When peripheral inflammation and/or nerve injury occur, transcription-dependent changes may ensue leading to longer-lasting effects [118] including, for example the expression of substance P and BDNF by neurons within the Dorsal Root Ganglia. Inflammation also exposes nerve terminals to nerve growth factor (NGF) [119, 120] which stimulates nociceptors expressing the tyrosine-kinase receptor TrkA a high-affinity receptor for NGF. These, when stimulated by NGF express higher levels of neuropeptides and other NGF-dependent proteins [121].
Once this process has occurred, low-intensity innocuous stimuli can now mediate the release of the aforementioned neuropeptides into the spinal cord [118]. This leads to an induction of cyclooxygenase-2 (Cox-2) in the dorsal horn neurons which then increases prostaglandin E2 (PGE2) production and release. When PGE2 binds to its dorsal horn neuron receptor, it elicits several changes including: potentiation of AMPAR and NMDAR currents, activation of nonselective cation channels, reduction in inhibitory glycinergic neurotransmission and an effect on EP4 receptors on presynaptic terminals resulting in an increase in neurotransmitter release [122]. In addition, spinal cord microglial cells also release pro-inflammatory cytokines including TNF and IL-1, which enhance excitatory and reduced inhibitory currents through COX-2 activation [123]. COX-2 has experimentally been suppressed in neurons, with a resulting near-complete loss of mechanical pain hypersensitivity but a retention of heat hyperalgesia in response to peripheral inflammation [122]. As mentioned above, mechanical sensitivity is a major feature of central sensitization [1], thus the COX-2 pathway’s importance has been demonstrated.
Peripheral inflammation can also elicit other important changes, such as a shift from GluR2/3 to GluR1-containing AMPARs in the dorsal root neurons of the superficial lamina [124–126]. This leads to transforming previously Ca2+ impermeable to Ca2 + -permeable AMPARs which then become a major source of the [Ca2+]i increase in inflammatory pain, generating as much Ca2+ influx as with NMDAR activation [127]. Additionally, the NMDAR becomes phosphorylated by protein kinase C (pPKC) and extracellular signal-regulated kinase 2, which increase receptor activity [128]. Peng et al. investigated the participation of cyclin-dependent kinase-5 (Cdk5)-mediating NMDAR NR2B subunit phosphorylation in cross-organ reflex sensitization caused by colon irritation. They used external urethral sphincter electromyogram (EUSE) reflex activity evoked by the pelvic afferent nerve test stimulation (TS, 1 stimulation/30 s) and measured protein expression in the spinal cord and dorsal root ganglion tissue (T13-L2 and L6-S2 ipsilateral to the stimulation) in response to colon mustard oil instillation in anesthetized rats. They found that when compared with a baseline reflex activity with a single action potential evoked by the TS before the administration of test agents, mustard oil instillation into the descending colon sensitized the evoked activity characterized by elongated firing in the reflex activity in association with increased protein levels of Cdk5, PSD95, and phosphorylated NR2B (pNR2B) but not of total NR2B (tNR2B) in the spinal cord tissue. Both the cross-organ reflex sensitization and increments in protein expression were reversed by intra-colonic pretreatments with ruthenium red (a nonselective TRPV, antagonist), capsaizepine (a TRPV1-selective antagonist), lidocaine (a nerve conduction blocker) as well as by the intra-thecal pretreatment with APV (a NRMDR antagonist) Co-101244 (a NR2B-selective antagonist) and roscovitine (a Cdk5 antagonist). Moreover, compared with the control group, both the increase in pNR2B and the cross-organ reflex sensitization were attenuated in the si-RNA of NR2B rats. These results suggested that Cdk-dependent NMDAR NR2B subunit phosphorylation mediates the development of cross-organ pelvic-urethra reflex sensitization caused by acute colon irritation. The authors suggest that this mechanism could possibly underlie the high concurrence of pelvic pain syndrome with IBS [67]. Similar findings have been made in using other colitis models where inflammation up-regulates the activity of NMDARs in DRG neurons within ganglia innervating this tissue through mechanisms involving increased expression and persistent tyrosine phosphorylation [129]. Finally, peripheral inflammation also promotes group I mGluR insertion into the membrane (mGluR5) in a location closer to the synapse (mGluR1), thereby further increasing receptor expression at the level of the synaptic terminal [130].
Related posts:
 Gastroparesis: Pathophysiology of Chronic Abdominal Pain and Current Treatment
Chronic Abdominal Pain of Gynecologic Causes: Diagnosis and Treatment
Gastric and Other Visceral Stimulation for Chronic Painful Gastrointestinal Motility Disorders
Gastroparesis: Pathophysiology of Chronic Abdominal Pain and Current Treatment
Chronic Abdominal Pain of Gynecologic Causes: Diagnosis and Treatment
Gastric and Other Visceral Stimulation for Chronic Painful Gastrointestinal Motility Disorders
 Establishing Diagnosis of Chronic Abdominal Pain: Gastroenterologist View
Establishing Diagnosis of Chronic Abdominal Pain: Gastroenterologist View
 Peripheral Nerve Stimulation for Chronic Abdominal Pain
Peripheral Nerve Stimulation for Chronic Abdominal Pain
 The Epidemiology of Chronic Abdominal Pain
The Epidemiology of Chronic Abdominal Pain
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


