Disorders of Glucose Homeostasis
Edward Vincent S. Faustino
Stuart A. Weinzimer
Michael F. Canarie
Clifford W. Bogue
KEY POINTS
 Diabetic ketoacidosis (DKA) is the most frequent cause of diabetes-related death in children, with a mortality rate of 0.2%-0.3%. Over 75%-87% of the mortality in DKA can be attributed to cerebral edema.
Diabetic ketoacidosis (DKA) is the most frequent cause of diabetes-related death in children, with a mortality rate of 0.2%-0.3%. Over 75%-87% of the mortality in DKA can be attributed to cerebral edema. Correction of dehydration with isotonic fluids and insulin replacement is necessary for optimal management of DKA. Insulin infusion should not be discontinued until DKA resolves. Glucose should be provided to avoid hypoglycemia.
Correction of dehydration with isotonic fluids and insulin replacement is necessary for optimal management of DKA. Insulin infusion should not be discontinued until DKA resolves. Glucose should be provided to avoid hypoglycemia. Early identification and prompt management of cerebral edema is crucial in improving the outcomes in children with DKA.
Early identification and prompt management of cerebral edema is crucial in improving the outcomes in children with DKA. Hyperglycemic hyperosmolar syndrome (HHS) is underrecognized in children and may be mistaken for DKA.
Hyperglycemic hyperosmolar syndrome (HHS) is underrecognized in children and may be mistaken for DKA. Early and aggressive fluid therapy is critical for effective management of HHS.
Early and aggressive fluid therapy is critical for effective management of HHS. Mortality in HHS is high, 15% or higher, attributable to circulatory collapse, thrombotic complications, and multisystem organ failure.
Mortality in HHS is high, 15% or higher, attributable to circulatory collapse, thrombotic complications, and multisystem organ failure. Hyperglycemia commonly occurs in critically ill nondiabetic children and is associated with increased morbidity and mortality.
Hyperglycemia commonly occurs in critically ill nondiabetic children and is associated with increased morbidity and mortality. Neurohormonal factors that accompany stress response lead to insulin resistance, increased glucose production, and hyperglycemia.
Neurohormonal factors that accompany stress response lead to insulin resistance, increased glucose production, and hyperglycemia. The optimal management of hyperglycemia in critically ill children is unclear. Controlling blood glucose to a target range of 80-110 mg/dL is likely not beneficial in postoperative cardiac children. The benefit of tight glycemic control in critically ill children with other conditions is uncertain.
The optimal management of hyperglycemia in critically ill children is unclear. Controlling blood glucose to a target range of 80-110 mg/dL is likely not beneficial in postoperative cardiac children. The benefit of tight glycemic control in critically ill children with other conditions is uncertain. Except in rare circumstances, hypoglycemia is a defect in fasting adaptation. Understanding the ordered responses to fasting provides the framework for differentiating among the various causes of hypoglycemia and making a prompt diagnosis.
Except in rare circumstances, hypoglycemia is a defect in fasting adaptation. Understanding the ordered responses to fasting provides the framework for differentiating among the various causes of hypoglycemia and making a prompt diagnosis. Infants, young children, and critically ill children may not present with typical adrenergic symptoms of hypoglycemia. A high index of suspicion should be maintained for these patient populations.
Infants, young children, and critically ill children may not present with typical adrenergic symptoms of hypoglycemia. A high index of suspicion should be maintained for these patient populations. Collection of the appropriate blood and urine specimens for determination of alternate fuels and hormones at the time of hypoglycemia provides the most efficient opportunity for determining the etiology of hypoglycemia.
Collection of the appropriate blood and urine specimens for determination of alternate fuels and hormones at the time of hypoglycemia provides the most efficient opportunity for determining the etiology of hypoglycemia. Although the outcome of hypoglycemia in a child probably depends on the underlying cause, the clinical significance of insulin-induced hypoglycemia in critically ill children is less clear. Follow-up studies on tight glycemic control trials in children will provide valuable information to answer this question.
Although the outcome of hypoglycemia in a child probably depends on the underlying cause, the clinical significance of insulin-induced hypoglycemia in critically ill children is less clear. Follow-up studies on tight glycemic control trials in children will provide valuable information to answer this question.DIABETIC KETOACIDOSIS IN CHILDREN
Diabetic ketoacidosis (DKA) is a life-threatening, preventable complication of diabetes mellitus that is characterized by inadequate insulin action, hyperglycemia, dehydration, electrolyte loss, metabolic acidosis, and ketosis. It is the most frequent cause of death in children with type 1 diabetes mellitus.  Type 1 diabetes is one of the most common chronic illnesses in children with an incidence of ˜3% in young people worldwide (1), and DKA is one of the most common reasons for admission to the PICU (2). DKA occurs in ˜15%-70% of children with diabetes at disease onset and in 1%-10% of children with a previous diagnosis of diabetes (3). DKA at diagnosis of type 1 diabetes occurs more commonly in very young children, children without a prior family history of type 1 diabetes, and in families from lower socioeconomic backgrounds. The risk of DKA in children with established type 1 diabetes is associated with infection, intercurrent illness, insulin pump malfunction, purposeful insulin omission, a history of prior poor metabolic control, concomitant psychiatric disease, peripubertal and adolescent girls, and lower socioeconomic background (3,4).
Type 1 diabetes is one of the most common chronic illnesses in children with an incidence of ˜3% in young people worldwide (1), and DKA is one of the most common reasons for admission to the PICU (2). DKA occurs in ˜15%-70% of children with diabetes at disease onset and in 1%-10% of children with a previous diagnosis of diabetes (3). DKA at diagnosis of type 1 diabetes occurs more commonly in very young children, children without a prior family history of type 1 diabetes, and in families from lower socioeconomic backgrounds. The risk of DKA in children with established type 1 diabetes is associated with infection, intercurrent illness, insulin pump malfunction, purposeful insulin omission, a history of prior poor metabolic control, concomitant psychiatric disease, peripubertal and adolescent girls, and lower socioeconomic background (3,4).
Type 1 diabetes is one of the most common chronic illnesses in children with an incidence of ˜3% in young people worldwide (1), and DKA is one of the most common reasons for admission to the PICU (2). DKA occurs in ˜15%-70% of children with diabetes at disease onset and in 1%-10% of children with a previous diagnosis of diabetes (3). DKA at diagnosis of type 1 diabetes occurs more commonly in very young children, children without a prior family history of type 1 diabetes, and in families from lower socioeconomic backgrounds. The risk of DKA in children with established type 1 diabetes is associated with infection, intercurrent illness, insulin pump malfunction, purposeful insulin omission, a history of prior poor metabolic control, concomitant psychiatric disease, peripubertal and adolescent girls, and lower socioeconomic background (3,4).Definition and Pathophysiology
DKA is defined as a blood glucose concentration >200 mg/dL, with ketonemia/ketonuria, and a venous pH <7.3 or bicarbonate <15 mEq/L (3). The severity of DKA is categorized on the basis of the degree of acidosis (mild: venous pH <7.3 or bicarbonate <15 mEq/L; moderate: pH <7.2 or bicarbonate <10 mEq/L; and severe: pH <7.1 or bicarbonate <5 mEq/L). The primary abnormality is insulin deficiency, which results in hyperglycemia by three mechanisms: increased gluconeogenesis, accelerated glycogenolysis, and impaired peripheral glucose utilization. Early in the progression of type 1 diabetes, the defects in peripheral glucose uptake predominate over the abnormalities in hepatic glucose production, such that postprandial glucose levels are elevated while fasting glucose levels
are normal. However, as insulin deficiency becomes progressively more severe and absolute, fasting hyperglycemia occurs. As serum glucose levels exceed the renal threshold of 180 mg/dL, an osmotic diuresis occurs, which results in the loss of extracellular water and electrolytes.
are normal. However, as insulin deficiency becomes progressively more severe and absolute, fasting hyperglycemia occurs. As serum glucose levels exceed the renal threshold of 180 mg/dL, an osmotic diuresis occurs, which results in the loss of extracellular water and electrolytes.
Although insulin deficiency is the primary defect, physiologic stress caused by acidosis and progressive dehydration, as well as coexistent infection or illness, stimulate the release of counter-regulatory hormones (glucagon, catecholamines, and cortisol), which further exacerbate hyperglycemia by increasing hepatic glucose production and further impairing peripheral glucose uptake. Counter-regulatory hormones, particularly epinephrine, also promote lipolysis and free fatty acid release through the activation of adipose tissue hormonesensitive lipase in adipocytes. Additionally, these hormones activate the hepatic β-oxidation of free fatty acids to ketone bodies, predominantly β-hydroxybutyrate and acetoacetate, which leads to ketoacidosis. Accumulation of ketoacids is the primary cause of the metabolic acidosis in DKA. Although acetone is also formed (causes a fruity odor to the breath), it does not contribute to the acidosis.
The increasing levels of hyperglycemia and acidosis contribute to a vicious cycle. Osmotic diuresis leads to intravascular volume depletion with reduction in renal blood flow and glomerular perfusion, limiting the body’s ability to excrete glucose and worsening the hyperglycemia. Likewise, progressive dehydration and acidosis further stimulate the release of counter-regulatory hormones, which accelerates the production of glucose and ketoacids (Fig. 106.1). Worsening dehydration then leads to poor peripheral tissue perfusion, with resultant increases in lactic acidosis. Abdominal pain and vomiting are a result of intestinal ileus from ketoacids and dehydration and prevent hydration with oral fluids. The metabolic acidosis leads to potassium transport out of cells into the plasma in exchange for hydrogen and excretion in the urine. Thus, patients with DKA develop a “total-body” deficiency of potassium (intra- and extracellular), which may not correlate with their serum level. Phosphate is affected similarly. A deficiency of 2,3-diphosphoglycerate, a phosphate-containing glycolytic intermediate in red blood cells that facilitates release of oxygen from hemoglobin, may also contribute to the development of lactic acidosis, complicating the ketoacidosis. As sodium and potassium are excreted in the urine with lactate and ketoacids, hyperchloremia occurs. Electrolyte deficits of up to 5-10 mEq/kg of sodium, 3-5 mEq/kg of potassium, and 0.5-1.5 mmol/kg of phosphate in DKA are not uncommon.
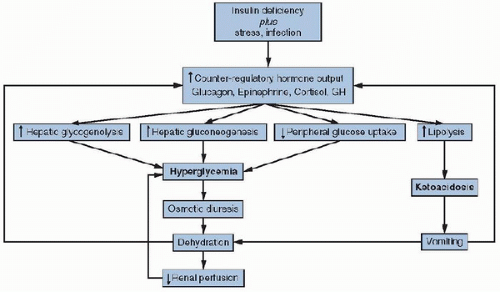 FIGURE 106.1. Pathophysiology of diabetic ketoacidosis. GH, growth hormone. |
Clinical Presentation and Differential Diagnosis
DKA is not difficult to recognize in a child with known diabetes who is dehydrated, hyperventilating, and obtunded. In the child whose diabetes has not yet been diagnosed, however, it may be confused with gastroenteritis, pneumonia, sepsis, toxic ingestion, or a central nervous system (CNS) lesion. The diagnosis of diabetes (if not already established) is suggested by a history of polyuria, polydipsia, polyphagia, nocturia, or enuresis in a previously toilet-trained child. Weakness and unexplained weight loss may also be presenting features. Abdominal pain, tenderness, and guarding are frequently present in DKA and may be of sufficient intensity to mimic an acute surgical abdomen. Other nonspecific symptoms of DKA, such as mental obtundation, vomiting, and abnormal breathing, are related to the dehydration and acidosis.
DKA in children must be differentiated from such commonly occurring childhood illnesses as urinary tract infection, gastroenteritis, more severe conditions that result in a “surgical abdomen,” asthma, and pneumonia. In all of these conditions, symptoms and signs may overlap with diabetes: urinary frequency, polyuria, nocturia, abdominal pain, vomiting, changes in breathing, and dehydration. In particular, extreme hyperglycemia, due to stress hormone excess, and acidosis, due to dehydration and/or fasting, may closely mimic DKA in infants and toddlers with a febrile illness associated with poor oral intake. However, in these children, the absence of polyuria, and polydipsia and the rapid clinical improvement with rehydration alone (without insulin) may differentiate these episodes of stress hyperglycemia from true DKA.
The physical examination findings in DKA are predominantly those of dehydration: tachycardia, delayed capillary refill, dry mucous membranes, and poor skin turgor. Severe acidosis and dehydration may impair cardiac contractility,
resulting in hypotension. Respiratory compensation for the metabolic acidosis induces hyperventilation, which may appear as deep sighing respirations (Kussmaul breathing) and may initially suggest a primary respiratory illness. Acetone on the breath yields a fruity odor and may be present but is not reliably detected. Mental status may vary from full alertness to frank coma. Most patients with severe metabolic derangements are lethargic but do not have severe CNS depression and are able to protect their airway.
resulting in hypotension. Respiratory compensation for the metabolic acidosis induces hyperventilation, which may appear as deep sighing respirations (Kussmaul breathing) and may initially suggest a primary respiratory illness. Acetone on the breath yields a fruity odor and may be present but is not reliably detected. Mental status may vary from full alertness to frank coma. Most patients with severe metabolic derangements are lethargic but do not have severe CNS depression and are able to protect their airway.
The primary survey should focus initially on the adequacy of the airway, breathing, a thorough assessment of the circulatory status (heart rate, blood pressure, description of mucous membranes, capillary refill, distal pulses, and warmth of extremities), degree of dehydration (including weight if possible), and mental status. A rough estimation of degree of dehydration should be made to calculate the fluid deficit and facilitate rehydration therapy. A careful examination and written documentation of the neurologic status is critical to serve as a baseline in case of deterioration in neurologic status later in therapy. General physical examination should include respiratory and abdominal examinations, as well as a search for an intercurrent illness that may have served as the precipitating factor for the episode of DKA.
Laboratory Findings
The laboratory evaluation of patients suspected to have DKA includes determination of the blood glucose, plasma or urinary ketones, serum electrolyte concentration, blood urea nitrogen (BUN), creatinine, osmolarity, baseline calcium and phosphorus, and, if infection is suspected, complete blood count and blood culture. Bedside determination of the blood glucose with a glucose-monitoring device and evaluation of the urine for glucose and ketones should be performed as quickly as possible, and treatment should be initiated without waiting for the results of the laboratory assessment to become available. A baseline blood gas measurement should also be made to determine the pH and PCO2. While venous blood gas measurements suffice in most episodes of DKA, an arterial blood gas measurement should be considered in patients who are suspected to have incomplete respiratory compensation or those with hemodynamic instability.
The initial blood glucose level is characteristically, but not invariably, elevated. Hyperglycemia beyond the renal threshold for glucose filtration indicates reduced glomerular filtration. Although the definition of DKA presupposes hyperglycemia, ketoacidosis with normal or even low glucose levels may occur in patients with known diabetes who have taken insulin recently. Poor oral intake or vomiting may also present with near-normal glucose concentrations.
Measurement of serum electrolytes in DKA reveals a low bicarbonate level and increased anion-gap metabolic acidosis, primarily due to the unmeasured ketoacids, including β-hydroxybutyrate and acetoacetate, as well as lactate in situations of severe tissue hypoxia. Most urine dipsticks that utilize a nitroprusside reaction to measure ketones detect only acetoacetate. In normal individuals, the ratio of β-hydroxybutyrate to acetoacetate is ˜1:1. However, this ratio may increase to 10:1 or more in DKA. Therefore, although useful to diagnose ketosis, these strips are not helpful in gauging the severity of DKA or response to treatment. Patients in DKA typically have an anion gap of 20-30 mEq/L. An anion gap >35 mEq/L suggests a concomitant lactic acidosis (3).
Serum electrolyte concentrations are almost always abnormal in DKA and may not accurately reflect total body electrolyte disturbances. Because of the osmotic flux of water from the intracellular space to the extracellular space in the presence of hyperglycemia, serum sodium will be reduced. The actual sodium concentration may be determined by adding 1.6 mEq/L for each 100 mg/dL rise in glucose concentration >100 mg/dL (5). Furthermore, extreme hypertriglyceridemia may cause the serum sodium concentration to be falsely lowered (dilutional hyponatremia) (6). A normal or elevated sodium level in the setting of severe DKA suggests extreme free water losses. The degree of elevation of the BUN and creatinine, as well as the hematocrit, may also indicate the extent of dehydration (and the possibility of renal damage).
The initial serum potassium may be low, normal, or high, depending on the degree of acidosis and the quantitative urinary losses. However, total body potassium stores are almost always depleted from excessive urinary losses, and normal or low K levels at presentation may be associated with severe hypokalemia once treatment for DKA is initiated. Phosphate, like potassium, is depleted in the setting of DKA, and serum phosphate levels may not accurately reflect total body stores. Serum phosphate level may also decrease during therapy.
Clinical Management
The International Society for Pediatric and Adolescent Diabetes published practice guidelines for the management of DKA in 2009 (3). The guidelines are consistent with those published  by the Lawson Wilkins Pediatric Endocrine Society and European Society for Paediatric Endocrinology in 2004 (7). The goals of therapy for DKA are (1) to correct dehydration and electrolyte deficits, (2) to correct acidosis and reverse ketosis, (3) to restore blood glucose to near normal, (4) to avoid complications of therapy, and (5) to identify and treat any precipitating event.
by the Lawson Wilkins Pediatric Endocrine Society and European Society for Paediatric Endocrinology in 2004 (7). The goals of therapy for DKA are (1) to correct dehydration and electrolyte deficits, (2) to correct acidosis and reverse ketosis, (3) to restore blood glucose to near normal, (4) to avoid complications of therapy, and (5) to identify and treat any precipitating event.
by the Lawson Wilkins Pediatric Endocrine Society and European Society for Paediatric Endocrinology in 2004 (7). The goals of therapy for DKA are (1) to correct dehydration and electrolyte deficits, (2) to correct acidosis and reverse ketosis, (3) to restore blood glucose to near normal, (4) to avoid complications of therapy, and (5) to identify and treat any precipitating event.Correction of Dehydration and Electrolyte Deficits
A rough estimation of degree of dehydration should be made to calculate the fluid deficit and facilitate rehydration therapy even though such clinical approximations are subjective and typically inaccurate in estimating the actual degree of dehydration. Furthermore, the dehydration of DKA is hyperosmolar, resulting in a translocation of intracellular water to the extracellular compartment, thus masking the degree of dehydration. Significant total body fluid losses can occur before evidence of changes in vital signs or physical examination. Serum BUN and hematocrit may be useful markers of dehydration. As in other forms of hypertonic dehydration, too rapid fluid administration should be avoided in children in DKA. The fluid deficit should be corrected gradually, over 48 hours. The rare DKA patient who is hemodynamically unstable at the presentation should receive a 10 mL/kg normal saline or Ringer’s lactate bolus. Only one additional 10 mL/kg isotonic fluid bolus should be given if hemodynamic instability has not resolved. Thereafter, inotropic support, invasive monitoring, and an echocardiogram are required to guide hemodynamic management.
Serial calculations of actual (as opposed to measured) serum sodium concentrations are recommended, as sodium levels that fail to rise with treatment may signify excessive free water accumulation and an increased risk of cerebral edema (8,9). It is unusual to provide fluids in excess of 1.5-2 times the daily maintenance rate. The initial fluid replacement (first 6 hours) generally utilizes isotonic fluids. Subsequent fluids should contain at least 0.45% saline to ensure a gradual decline in serum osmolarity and prevent excessive free water accumulation (10). Hyperchloremic metabolic acidosis may develop with continued use of large volumes of normal saline. There are no data to support the use of colloids over crystalloids for this patient group (3).
Early potassium replacement is important to correct the deficit. With the initiation of insulin therapy and correction
of acidosis, serum potassium levels may drop precipitously as potassium shifts back from the extracellular to the intracellular compartment. In hypokalemic patients, replace potassium at the time of initial volume expansion and prior to starting insulin infusion. Otherwise, potassium should be administered only after adequate renal function is determined, particularly in patients who are hyperkalemic. Electrocardiographic monitoring facilitates early recognition of hyperkalemia (peaked T waves), hypokalemia (flat or inverted T waves), and the development of potentially dangerous cardiac dysrhythmias. Potassium is usually given at a dose of 40 mEq/L of fluid, either as potassium chloride or in combination with potassium phosphate. The latter has the additional advantage of replacing the phosphate deficit. Potassium can also be repleted as potassium acetate, which may theoretically provide buffering equivalents for the metabolic acidosis. The relative risks and benefits of these infusion alternatives in the treatment of DKA in children have not been rigorously studied (3,7). It should be noted that while hypophosphatemia in DKA may predispose to rhabdomyolysis and hemolysis and while low 2,3-diphosphoglycerate levels in DKA may impair tissue delivery of oxygen, complications of hypophosphatemia in DKA are rare, and phosphate replacement may precipitate acute hypocalcemia. Serum calcium should be monitored if phosphate is given. It is recommended to replace phosphate in patients with severe hypophosphatemia and unexplained weakness (3).
of acidosis, serum potassium levels may drop precipitously as potassium shifts back from the extracellular to the intracellular compartment. In hypokalemic patients, replace potassium at the time of initial volume expansion and prior to starting insulin infusion. Otherwise, potassium should be administered only after adequate renal function is determined, particularly in patients who are hyperkalemic. Electrocardiographic monitoring facilitates early recognition of hyperkalemia (peaked T waves), hypokalemia (flat or inverted T waves), and the development of potentially dangerous cardiac dysrhythmias. Potassium is usually given at a dose of 40 mEq/L of fluid, either as potassium chloride or in combination with potassium phosphate. The latter has the additional advantage of replacing the phosphate deficit. Potassium can also be repleted as potassium acetate, which may theoretically provide buffering equivalents for the metabolic acidosis. The relative risks and benefits of these infusion alternatives in the treatment of DKA in children have not been rigorously studied (3,7). It should be noted that while hypophosphatemia in DKA may predispose to rhabdomyolysis and hemolysis and while low 2,3-diphosphoglycerate levels in DKA may impair tissue delivery of oxygen, complications of hypophosphatemia in DKA are rare, and phosphate replacement may precipitate acute hypocalcemia. Serum calcium should be monitored if phosphate is given. It is recommended to replace phosphate in patients with severe hypophosphatemia and unexplained weakness (3).
Correction of Acidosis and Reversal of Ketosis
Acidosis in DKA reverses with fluid replacement and insulin therapy. Restoration of circulation improves renal perfusion, resulting in acid excretion. Improved tissue perfusion also limits the production of lactic acid in hypoperfused tissues. Insulin administration stops ketoacid production. Acidosis in DKA usually resolves later than hyperglycemia. However, it is important to continue insulin therapy until acidosis resolves while supporting the patient with glucose to prevent hypoglycemia.
Bicarbonate, once considered routine therapy in severe DKA, is no longer thought to be beneficial and is no longer recommended in the treatment of uncomplicated DKA. Most often, dramatic clinical improvement results simply from initial expansion of extracellular fluid volume, reestablishment of adequate peripheral perfusion, and prompt insulin administration. Bicarbonate therapy may lead to paradoxical worsening of CNS acidosis, hypokalemia, sodium overload, late alkalosis, and, theoretically, impaired tissue perfusion due to leftward shift of the oxyhemoglobin dissociation curve. Finally, bicarbonate has been associated, in multivariate analysis, with a higher risk of development of cerebral edema (8). Bicarbonate may still be considered in the setting of severe acidosis with impaired cardiac contractility and vasomotor tone. It may be given in a slow infusion of 1-2 mEq/kg over 60 minutes.
Restoration of Blood Glucose to Near Normal
Insulin therapy, at a dose of 0.05-0.1 U/kg/h, should be initiated immediately after the patient has received initial volume expansion. Because insulin adsorbs to intravenous tubing, 20 mL of the insulin solution should be flushed through the tubing set before connecting to the patient. The insulin infusion can then be started immediately, and there is no need to allow the insulin to “dwell” in the intravenous tubing after the 20-mL flush (11). Intravenous insulin bolus is contraindicated because it may increase the risk of cerebral edema. As with fluid replacement, the aim of therapy is gradual correction of blood glucose (i.e., a reduction of the blood glucose by 50-100 mg/dL/h). However, the serum glucose concentration often falls significantly with initial rehydration alone owing to increased glomerular filtration from improved renal perfusion. The dose of insulin usually remains at 0.1 U/kg/h until acidosis resolves. Because insulin infusion often results in lowering of the blood glucose concentration before the decrease in ketoacidosis, it is important not to decrease the rate of insulin infusion, but instead to add dextrose to the rehydration fluids to prevent hypoglycemia. Dextrose should be added to the intravenous fluid solution when the serum glucose level falls below 250 mg/dL and should then be titrated to maintain blood glucose at about 200 mg/dL until DKA resolves. This may be accomplished with the simultaneous use of two intravenous solutions that differ only in the dextrose concentration (10% and 0%). Independent manipulation of each infusion allows the dextrose infusion to be varied quickly and efficiently (the so-called “two-bag” system) (12).
Avoidance of Complications of Therapy
Some of the more common complications of DKA therapy are inadequate rehydration, hypoglycemia, hypokalemia, hyperchloremic  acidosis, and cerebral edema. Of these, acute cerebral edema is the most serious, and accounts for 75%-87% of the mortality in DKA. Cerebral edema occurs in ˜0.5%-1% of episodes of pediatric DKA and carries a mortality rate of over 20% and a rate of permanent neurologic disability of over 25% (13). Cerebral edema is more likely to occur in patients at the time of first diagnosis of diabetes, in younger children, and in those presenting with the most severe degree of dehydration and metabolic derangement. It typically develops within the first 4-12 hours of treatment but may occur as late as 24-48 hours after the start of treatment. Symptoms and signs include headache, confusion, slurred speech, bradycardia, hypertension, and other signs of increased intracranial pressure. A bedside scoring system based on the patient’s neurologic status was proposed to diagnose cerebral edema (14). Although a number of causes of acute cerebral edema have been suggested from uncontrolled, retrospective studies, the etiology in many cases is not known. A common proposed mechanism involves a rapid decline in serum osmolality with treatment of DKA. On the basis of this hypothesis, previous recommendations for treatment of DKA advised limiting the rate of rehydration to <4 L/m2/day (15) and avoiding hypotonic fluids (16). Until the precise mechanisms that underlie the development and progression of cerebral edema are elucidated, it appears prudent to correct dehydration evenly over 48 hours (unless the patient is in shock), and monitor neurologic status and laboratory parameters closely.
acidosis, and cerebral edema. Of these, acute cerebral edema is the most serious, and accounts for 75%-87% of the mortality in DKA. Cerebral edema occurs in ˜0.5%-1% of episodes of pediatric DKA and carries a mortality rate of over 20% and a rate of permanent neurologic disability of over 25% (13). Cerebral edema is more likely to occur in patients at the time of first diagnosis of diabetes, in younger children, and in those presenting with the most severe degree of dehydration and metabolic derangement. It typically develops within the first 4-12 hours of treatment but may occur as late as 24-48 hours after the start of treatment. Symptoms and signs include headache, confusion, slurred speech, bradycardia, hypertension, and other signs of increased intracranial pressure. A bedside scoring system based on the patient’s neurologic status was proposed to diagnose cerebral edema (14). Although a number of causes of acute cerebral edema have been suggested from uncontrolled, retrospective studies, the etiology in many cases is not known. A common proposed mechanism involves a rapid decline in serum osmolality with treatment of DKA. On the basis of this hypothesis, previous recommendations for treatment of DKA advised limiting the rate of rehydration to <4 L/m2/day (15) and avoiding hypotonic fluids (16). Until the precise mechanisms that underlie the development and progression of cerebral edema are elucidated, it appears prudent to correct dehydration evenly over 48 hours (unless the patient is in shock), and monitor neurologic status and laboratory parameters closely.
acidosis, and cerebral edema. Of these, acute cerebral edema is the most serious, and accounts for 75%-87% of the mortality in DKA. Cerebral edema occurs in ˜0.5%-1% of episodes of pediatric DKA and carries a mortality rate of over 20% and a rate of permanent neurologic disability of over 25% (13). Cerebral edema is more likely to occur in patients at the time of first diagnosis of diabetes, in younger children, and in those presenting with the most severe degree of dehydration and metabolic derangement. It typically develops within the first 4-12 hours of treatment but may occur as late as 24-48 hours after the start of treatment. Symptoms and signs include headache, confusion, slurred speech, bradycardia, hypertension, and other signs of increased intracranial pressure. A bedside scoring system based on the patient’s neurologic status was proposed to diagnose cerebral edema (14). Although a number of causes of acute cerebral edema have been suggested from uncontrolled, retrospective studies, the etiology in many cases is not known. A common proposed mechanism involves a rapid decline in serum osmolality with treatment of DKA. On the basis of this hypothesis, previous recommendations for treatment of DKA advised limiting the rate of rehydration to <4 L/m2/day (15) and avoiding hypotonic fluids (16). Until the precise mechanisms that underlie the development and progression of cerebral edema are elucidated, it appears prudent to correct dehydration evenly over 48 hours (unless the patient is in shock), and monitor neurologic status and laboratory parameters closely.Treatment of cerebral edema is aimed at lowering intracranial pressure. Reducing the rate of fluid administration by one-third is suggested. Prompt administration of IV mannitol (0.5-1 g/kg over 20 minutes) may be beneficial if given early in the course of cerebral edema. The dose may be repeated in 0.5-2 hours if there is no response to the initial dose. Hypertonic saline (3%), 5-10 mL/kg over 30 minutes, may be used as an alternative or in addition to mannitol. One should keep the patient’s head elevated and midline. Tracheal intubation may be needed for impending respiratory failure or airway protection. Although aggressive hyperventilation should be avoided as this decreases cerebral perfusion, care should be taken to match the level of hyperventilation the patient was spontaneously producing as sedating and ventilating the patient at a lower PaCO2 than they were producing may worsen acidosis, cause cerebral vasodilation, and increase intracranial pressure if intracranial compliance is limited. Near infrared spectroscopy (NIRS) may be used to assess the ventilatory parameters and resulting PaCO2 associated with a favorable cerebral O2 supply/demand ratio. Intracranial imaging to exclude other pathologies, such as cerebral infarction or thrombosis, should be obtained but not at the expense of timely therapeutic interventions.
Other less common complications of DKA include thrombosis (including cerebral venous sinus thrombosis), a concern in children who require a central venous catheter for access; cardiac dysrhythmias, usually related to electrolyte disturbances; pulmonary edema; renal failure; pancreatitis; rhabdomyolysis; and infection, such as aspiration pneumonia, sepsis, and mucormycosis.
Identification and Treatment of Any Precipitating Event
Infection is a common precipitating event for DKA. Infections should be appropriately worked up and treated. In cases of DKA in children with known diabetes, additional education on insulin omission, or sick day or pump failure management may be needed to prevent another episode of DKA.
Successful management of DKA requires meticulous attention to clinical and laboratory changes. Children with severe DKA or who are at increased risk for cerebral edema should be cared for in the ICU. Clinical monitoring is essential. Vital signs, perfusion, input/output balance, and neurologic status should be documented at least hourly. Cardiac monitoring is recommended owing to the risk of dysrhythmia. Laboratory monitoring should include venous blood glucose concentrations hourly and electrolytes and venous pH every 2-4 hours until normal. BUN and creatinine levels should be followed until they are normal, and calcium should be monitored, particularly if phosphate is administered. Lack of improvement in clinical and biochemical parameters with time suggests an occult infection or inadequate insulin or fluid replacement.
Conclusions and Future Directions—Diabetic Ketoacidosis
One of the most important areas for further investigation in the management of DKA is in improving prevention strategies. DKA, especially in known diabetics, is preventable. Future research endeavors should also address the basic pathophysiologic mechanisms of cerebral edema, so that treatment algorithms may be optimized to reduce or eliminate this complication of DKA. Differentiation of hyperosmolar coma from DKA and management of DKA in children with type 2 diabetes are areas that require more attention, as increasing numbers of pediatric patients with type 2 diabetes are being recognized. Finally, the use of continuous glucose-monitoring systems that track glucose values and trends in real time to facilitate clinical management of DKA have yet to be fully explored.
HYPERGLYCEMIC HYPEROSMOLAR SYNDROME
Hyperglycemic hyperosmolar syndrome (HHS) is a potentially lethal disorder of decompensated glucose homeostasis. Patients with this syndrome, known formerly as hyperosmolar nonketotic coma or state (HONKC) or hyperglycemic hyperosmolar nonketotic syndrome (HHNS), suffer from the dangerous consequences of marked hyperglycemia and hyperosmolarity. Once thought limited to the adult population, the  syndrome is increasingly described in younger patients.
syndrome is increasingly described in younger patients.
syndrome is increasingly described in younger patients.Epidemiology
Cases of extreme hyperglycemia without significant ketosis have been described in adults for more than 100 years (17). Currently, the literature cites the rate of HHS as 17.5 per 100,000 adult patients, with a greater distribution in the elderly and disabled (18). The syndrome affects known diabetics (particularly those with type 2 diabetes) but may also herald its onset. In addition, precipitating factors associated with HHS include infections, coexisting medical problems (e.g., renal failure, pancreatitis), medications (notably, diuretics, steroids, anticonvulsants, and psychotropic drugs), and total parenteral nutrition, as well as other conditions that lead to dehydration, such as burns and heat stroke (18).
HHS was first described in children in 1951, followed by sporadic case reports over the succeeding decades involving patients from infancy through adolescence (19). Often depicted as idiopathic, various triggers akin to those in adults have been ascribed to the syndrome in children. In addition, concerns about an underlying incipient diabetes mellitus (“pseudodiabetes of infancy”) or impaired osmoregulatory function have been considered (20). Unfortunately, the lack of data has precluded any reasonable attempt at estimating the syndrome’s prevalence in the pediatric population. Nonetheless, a review of published HHS cases in the past decade reveals an increasing number of reports (65 between 2001 and 2008), compared with the roughly 2 dozen reports described in the preceding decade (21,22,23,24,25). In contrast to the various etiologies described in the past, more recent reports noted a near-universal association with diabetic disorders (type 1 and 2 diabetes mellitus). Interestingly, among survivors of HHS, 41% had type 2 diabetes, while 35% had type 1 diabetes. Three-quarters of the patients were obese. Moreover, the risk appears higher in certain ethnic groups, with African Americans representing 70% of pediatric patients (25). This is consistent with the higher risk of type 2 diabetes in African Americans. The epidemic of pediatric obesity and its strong association with altered glucose metabolism, insulin resistance, and, ultimately, type 2 diabetes creates the potential for an increase in the number of cases of HHS in children (26,27).
Pathophysiology
HHS and DKA are often depicted as opposite extremes along the spectrum of decompensated diabetes. In HHS, insulin levels may be sufficient to suppress lipolysis and ketogenesis seen in DKA, but they are inadequate to promote normal anabolic function and inhibit the creation of glucose through gluconeogenesis and glycogenolysis. Thus, HHS may be seen as the presentation of an unrecognized relative insulin deficiency that is triggered by intercurrent illness or medication. The hyperbolic shape of the insulin sensitivity curve may help to explain the seemingly acute decompensation. Regardless of the proximal cause, the resulting surge of counter-regulatory hormones raises glucose levels by both enhanced hepatic glucose generation and worsening insulin resistance. Hyperglycemia itself triggers a heightened inflammatory state, which, in turn, exacerbates glucose dysregulation (28




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
