INTRODUCTION AND EPIDEMIOLOGY
Diabetic ketoacidosis (DKA) is an acute, life-threatening complication of diabetes mellitus. DKA occurs predominantly in patients with type 1 (insulin-dependent) diabetes mellitus, but 10% to 30% of cases occur in newly diagnosed type 2 (non–insulin-dependent) diabetes mellitus, especially in African Americans and Hispanics.1,2 Between 1993 and 2003, the yearly rate of U.S. ED visits for DKA was 64 per 10,000 with a trend toward an increased rate of visits among the African American population compared with the Caucasian population.3 Europe has a comparable incidence. A better understanding of the pathophysiology of DKA and an aggressive, uniform approach to its diagnosis and management have reduced mortality to <5% of reported episodes in experienced centers.4 However, mortality is higher in the elderly due to underlying renal disease or coexisting infection and in the presence of coma or hypotension.
PATHOPHYSIOLOGY
Figure 225-1 illustrates the complex relationships between insulin and counterregulatory hormones. DKA is a response to cellular starvation brought on by relative insulin deficiency and counterregulatory or catabolic hormone excess (Figure 225-1). Insulin is the only anabolic hormone produced by the endocrine pancreas and is responsible for the metabolism and storage of carbohydrates, fat, and protein. Counterregulatory hormones include glucagon, catecholamines, cortisol, and growth hormone. Complete or relative absence of insulin and the excess counterregulatory hormones result in hyperglycemia (due to excess production and underutilization of glucose), osmotic diuresis, prerenal azotemia, worsening hyperglycemia, ketone formation, and a wide-anion-gap metabolic acidosis.4
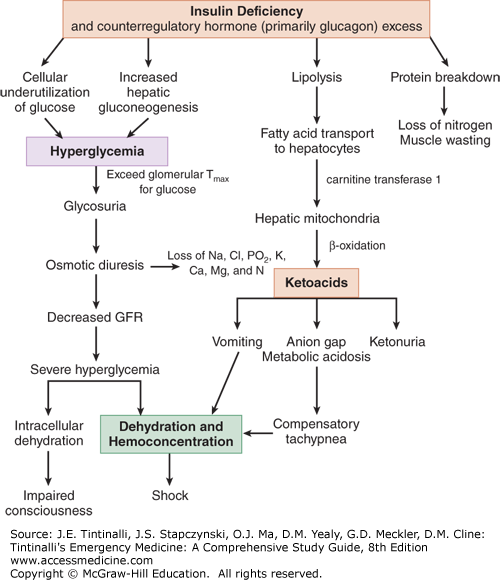
Ingested glucose is the primary stimulant of insulin release from the β cells of the pancreas. Insulin’s main action occurs at the three principal tissues of energy storage and metabolism—the liver, adipose tissue, and skeletal muscle. Insulin acts on the liver to facilitate the uptake of glucose and its conversion to glycogen while inhibiting glycogen breakdown (glycogenolysis) and suppressing gluconeogenesis. The net effect of these actions is to promote the storage of glucose in the form of glycogen. Insulin increases lipogenesis in the liver and adipose cells by producing triglycerides from free fatty acids and glycerol while inhibiting the breakdown of triglycerides. Insulin stimulates the uptake of amino acids into muscle cells with subsequent incorporation into muscle protein while preventing the release of amino acids from muscle and hepatic protein sources.
Deficiency in insulin secretion due to loss of islet cell mass is the predominant defect in type 1 diabetes mellitus. In the initial stages of diabetes mellitus, the secretory failure of β cells impairs fuel storage and may be evident only during a glucose tolerance test. As levels of insulin decrease, fuel stores are mobilized during fasting, resulting in hyperglycemia. When pancreatic β-cell reserve is present, hyperglycemia may trigger an increase in insulin and a return to normal glucose concentration. With further disease progression, hyperglycemia can no longer trigger an increase in insulin activity. Despite the presence of elevated intravascular glucose, in the absence of insulin, cells are unable to use glucose as a fuel source. The body responds by breaking down protein and adipose stores to try to produce a usable intracellular fuel. Loss of the normal physiologic effects of insulin leads to secretion of catabolic (counterregulatory) hormones and resulting hyperglycemia and ketonemia.
The response to cellular starvation seen with insulin insufficiency is increased levels of glucagon, catecholamines, cortisone, and growth hormone. Glucagon is the primary counterregulatory hormone. The catabolic effects of these hormones include increased gluconeogenesis and glycogenolysis, breakdown of fats into free fatty acids and glycerol, and proteolysis with increased levels of amino acids. Increased levels of glucogenic precursors, such as glycerol and amino acids, facilitate gluconeogenesis, worsening hyperglycemia.
Free fatty acids released in the periphery are bound to albumin and transported to the liver, where they undergo conversion to ketone bodies. The primary ketone bodies β-hydroxybutyrate (βHB) and acetoacetic acid (AcAc) account for the metabolic acidosis seen in DKA. The two are in equilibrium: AcAc + NADH βHB + NAD. AcAc is metabolized to acetone, another major ketone body. Depletion of hepatic glycogen stores favors ketogenesis. Low or absent insulin levels decrease the ability of the brain and cardiac and skeletal muscle to use ketones as an energy source, increasing ketonemia. The persistently elevated serum glucose level eventually causes an osmotic diuresis. The resulting volume depletion worsens hyperglycemia and ketonemia.
The renin-angiotensin-aldosterone system, activated by volume depletion, exacerbates renal potassium losses already occurring from osmotic diuresis. In the kidney, chloride is retained in exchange for the ketoanions being excreted. The loss of ketoanions represents a potential loss of bicarbonate. In the face of marked ketonuria, a superimposed hyperchloremic acidosis is also present. As adipose tissue is broken down, prostaglandins I2 and E2 are produced. Both account for paradoxical vasodilation that occurs despite profound levels of volume depletion.
Factors known to precipitate DKA are listed in Table 225-1.4 Additional risk factors include poor economic background, lack of insurance or minority status, drug abuse, depression, and the presence of an eating disorder. In many patients, no clear precipitating cause is found.4
Omission or reduced daily insulin injections Dislodgement/occlusion of insulin pump catheter Infection Pregnancy Hyperthyroidism, pheochromocytoma, Cushing’s syndrome Substance abuse (cocaine) Medications: steroids, thiazides, antipsychotics, sympathomimetics Heat-related illness Cerebrovascular accident GI hemorrhage Myocardial infarction Pulmonary embolism Pancreatitis Major trauma Surgery |
CLINICAL FEATURES
The clinical manifestations of DKA are related directly to hyperglycemia, volume depletion, and acidosis. The metabolic alterations of DKA tend to evolve within 24 hours.4 Osmotic diuresis gradually leads to volume loss in addition to renal losses of sodium, chloride, potassium, phosphorous, calcium, and magnesium. Initially, patients may compensate by increasing fluid intake, and polyuria and polydipsia are usually the only symptoms until ketonemia and acidosis develop. As acidosis progresses, ventilation is stimulated physiologically by acidemia to diminish the PCO2 and to counter metabolic acidosis. Acidosis combined with the effects of prostaglandins I2 and E2 leads to peripheral vasodilation despite profound levels of volume depletion. Prostaglandin release is also felt to play a role in unexplained nausea, vomiting, and abdominal pain that are seen frequently at presentation, especially in children. Vomiting, which may be a maladaptive physiologic response to diminish the acid load, unfortunately exacerbates potassium losses. As volume depletion progresses, poor absorption of SC insulin renders its administration ineffective. Impaired mental status may develop and is most likely multifactorial, related to metabolic acidosis, hyperosmolarity, low extracellular fluid volume, and poor hemodynamics. Alteration of consciousness has been reported to correlate better with elevated serum osmolality (>320 mOsm/L or >320 mmol/kg) than with severity of metabolic acidosis.5
Tachycardia, orthostasis or hypotension, poor skin turgor, and dry mucous membranes result from volume depletion. Kussmaul respirations, increased rate and depth of breathing, may be observed. Acetone produces the characteristic fruity odor on the breath found in some patients. The absence of fever does not exclude infection. Hypothermia is present occasionally because of peripheral vasodilation.
Abdominal pain and tenderness associated with DKA generally correlates with the level of acidosis. Pain can be due to gastric dilatation, ileus, or pancreatitis, but any other acute abdominal disorder can also develop. Due to the frequency of abdominal pain and the presence of an elevated serum amylase or lipase level in both DKA and pancreatitis, distinguishing these two conditions may be difficult. An elevated serum lipase level is more specific to pancreatitis, but it may also be elevated in DKA.
DIAGNOSIS
A blood glucose level >250 milligrams/dL (13.8 mmol/L), an anion gap >10 mEq/L (>10 mmol/L), a bicarbonate level <15 mEq/L (<15 mmol/L), and pH <7.3 with moderate ketonuria or ketonemia constitute the diagnosis of DKA.4,6 Patients may sometime present with “euglycemic ketoacidosis” (glucose <300 milligrams/dL or <16.6 mmol/L), such as those who present just after receiving insulin, type 1 diabetics who are young and vomiting, patients with impaired gluconeogenesis (alcohol abuse or liver failure), patients with low caloric intake/starvation, patients with depression, or pregnant patients. Euglycemic DKA has recently been described as an adverse effect of sodium-glucose cotransporter 2 (SGLT-2) inhibitors (canaglifozin and others). Check blood ketones, venous pH, bicarbonate levels and anion gap) to avoid missing euglycemic DKA, because the degree of hyperglycemia may not be significant (Table 225-2).7
Patients presenting shortly after receiving insulin Type 1 diabetics who are young and vomiting Patients with impaired gluconeogenesis (alcohol abuse or liver failure) Low caloric intake/starvation Depression Pregnancy |
The differential diagnosis of DKA (Table 225-3) includes any cause of a high anion gap metabolic acidosis. Patients with hyperosmolar, nonketotic coma tend to be older, have a more prolonged course, and have prominent mental status changes. Serum glucose levels generally are much higher (>600 milligrams/dL or >33.3 mOsm/L), and there is little to no anion gap metabolic acidosis.4 The ketosis in alcoholic ketoacidosis and starvation ketosis tend to be milder, and the serum glucose level is usually low or normal. βHB predominates in alcoholic ketoacidosis, so the urinary ketone test may be negative or trace positive.
If an ingestion cannot be excluded, serum osmolarity or drug-level testing is required. Renal failure, anion gap acidosis, and liver function abnormalities may be due to acetaminophen toxicity. Depending on the hemodynamic status, lactic acidosis (poor perfusion) may occur simultaneously with DKA; in these cases, determination of the serum lactate level is indicated. Patients on metformin with new-onset renal insufficiency are at risk for developing type B (aerobic) lactic acidosis.
Obtain a rapid bedside glucose determination, a urine test strip, and an electrocardiogram to check for hyperkalemia, and obtain a CBC, serum electrolytes, BUN and creatinine, urinalysis, venous blood gas, and phosphate/magnesium/calcium levels. Calculate the anion gap. Blood cultures and other laboratory tests should be done as clinically indicated. Arterial blood gas determinations are optional but may be required for the diagnosis and monitoring of critically ill patients.
In DKA, elevated serum levels of βHB and AcAc cause acidosis and ketonuria (ketonemia). The nitroprusside reagent normally used to detect urine and serum ketones only detects AcAc; acetone is only weakly reactive and βHB not at all. Gas chromatography can be used to detect serum acetone but is expensive and time-consuming.
NADH accumulation in mitochondria, as may occur with lactic acidosis or alcohol metabolism, favors the βHB side of equilibrium noted earlier (AcAc + NADH βHB + NAD). Paradoxically, as the patient is being treated and clinically improves, ketone levels will increase as the body converts the more acidic βHB to AcAc. Therefore, the blood ketone test (Acetest®) that uses the nitroprusside reaction is not a reliable measure for diagnosis or monitoring of DKA. Although not widely available, a new point-of-care capillary blood ketone test for βHB levels may be more sensitive and specific than traditional testing for urine ketones.6,8 As point-of-care capillary blood ketone testing for βHB becomes more widespread, this test may offer an alternative approach for the diagnosis and monitoring of DKA management compared to traditional measurements of serum HCO3, anion gap, and pH.9
DKA leads to a wide-anion-gap metabolic acidosis. Hyperchloremic acidosis also occurs on the basis of ketoanion exchange for chloride in the urine and is especially common in patients who maintain good hydration status and glomerular filtration rate despite ketoacidosis. Metabolic alkalosis may occur secondary to vomiting, osmotic diuresis, and concomitant diuretic use. Rarely, some patients with DKA may present with normal-appearing [HCO3–] or even an elevated [HCO3–], if coexisting metabolic alkalosis is severe enough to mask the acidosis. In such situations, an elevated anion gap may be the only clue to the presence of an underlying metabolic acidosis otherwise masked by the concomitant volume contraction–related metabolic alkalosis.
Venous pH has essentially replaced arterial blood gases in the assessment of the acid-base status of the DKA patient. A strong correlation exists between venous and arterial pH in patients with DKA, and the arterial blood gas value does not impact therapy.10 Venous pH is about 0.03 lower than arterial pH. Venous pH obtained during routine phlebotomy should be used to avoid arterial puncture, which is painful and may cause arterial vascular complications.
A low PCO2 determination usually reflects respiratory compensation for metabolic acidosis. If it is lower than explained by the degree of acidosis, a primary respiratory alkalosis exists, which may be an early indication of pulmonary disease (e.g., pneumonia, pulmonary embolus) or sepsis as a possible trigger of DKA. Chapter 15, Acid-Base Disorders, details how compensatory changes in PCO2 can be distinguished from a primary respiratory alkalosis.
Total-body potassium is depleted by renal losses. However, the measured serum potassium level is normal or elevated in most patients4 because of two important factors: extracellular shift of potassium secondary to acidemia and increased intravascular osmolarity caused by hyperglycemia. Although the actual incidence of initial hypokalemia in DKA is not known, a few studies report an occurrence of 4% to 6%.5,11,12 The decrease in serum potassium during therapy is reported to be about 1.5 mEq/L (1.5 mmol/L) and parallels the drop in glucose and the dose of insulin.5
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


