1. Pediatric responses to drugs are determined by a large number of factors that change independently of one another during growth and development.
2. Children aged less that 12 months may have greater absorption and distribution while the capacity for elimination is often reduced leading to an increased risk of overdose and toxicity.
3. Size, maturation and organ function are the key covariates explaining pharmacokinetic differences between pediatric patients and adults. Allometric theory can be used to explain the effects of size.
4. The greater volume of extracellular fluid (ECF) in infants is important as polar drugs such as aminoglycosides and neuromuscular blocking drugs distribute rapidly into ECF but enter cells more slowly. As a result, the initial dose of some drugs may be greater in infants than in children and adults (e.g. succinylcholine).
5. The concentrations and activities of many enzymes responsible for drug clearance are reduced or absent in newborns. Maturation may take from a few weeks to several years.
6. The interaction between a drug and its receptor make may be influenced by developmental changes in receptor number, type, affinity and the availability of natural ligands. In addition, age-related changes in organ size and blood flow impact on drug concentration at the effect site. Such changes have been implicated in altered responses to anesthetic drugs in the newborn.
7. The neuromuscular junction of the human neonate is three times more sensitive to the effects of nondepolarizing muscle relaxants than that of the adult due to a lack of acetylcholine in developing motor nerves. However, this sensitivity is countered by an increased volume of distribution, such that dose does not vary significantly with age.
8. The relative proportions of μ, κ, and δ opioid receptors and the selectivity of the μ receptor for its specific ligands changes with age. Such changes in the μ and κ receptors could be a factor in the increased susceptibility of newborns to the respiratory effects of opioids.
9. Lower concentrations of α1-acid glycoprotein in young infants increase the amount of unbound local anesthetic and the potential for CNS and cardiovascular toxicity.
10. Uptake and elimination of inhaled anesthetics occurs more rapidly in infants and young children than in adults due to an increased level of ventilation in relation to functional residual capacity, increased cardiac output and, in newborns, a reduced solubility of volatile agents in blood.
CHILDREN, ESPECIALLY INFANTS, differ from adults in the way they respond to drugs. These differences became apparent in the 1950s following a number of adverse drug events, including fatal cardiovascular collapse in infants treated with chloramphenicol (1) and an increase in kernicterus among newborns treated with sulfonamides (2). Pediatric responses to drugs are determined by a large number of factors that change independently of one another during growth and development.
The factors determining pediatric responses to drugs can be divided into those affecting pharmacokinetics (what the body does to the drug) and those affecting pharmacodynamics (what the drug does to the body). The aim of this chapter is to review these factors and describe how they affect the pharmacology of selected anesthetic drugs in infants and children.
BASIC PHARMACOKINETICS
Pharmacokinetics involves modeling the relationship between the plasma concentration of a drug and time and deriving various pharmacokinetic parameters (e.g., clearance and volume of distribution). In the simplest model, the body is considered to be a single compartment into which the drug is rapidly and evenly distributed. The relationship between plasma concentration and time may be represented by the following exponential equation:
![]()
In equation 2.1, Ct is the concentration of the drug at time t, Co is the initial plasma concentration, and k is the elimination rate constant. The apparent volume of distribution (Vd) of the drug is calculated by dividing the total dose by Co. The half-time (T1/2) is the time taken for the concentration of the drug to decline to one half of its original value. For an exponential process, T1/2 is constant and related to the elimination rate constant as follows:
![]()
CLINICAL PEARL Half-time does not predict dosing schedule; that is predicted by effect duration.
Plasma clearance (CL) is the volume of plasma cleared of the drug in unit time. It can be calculated thus:
![]()
By combining equations 2.2 and 2.3, the following useful relationship can be derived:
![]()
A single compartment model is often insufficient to describe the kinetics of drugs used in anesthesia, and a two- or even three-compartment model will be required. Two-compartment models can be interpreted using the plasma concentration–time graph that has two phases as shown in Figure 2.1. The initial steep decline (α phase) corresponds mainly to drug distribution from a central to a peripheral compartment, while the subsequent gradual decline (β phase) corresponds mainly to drug elimination. Plasma concentration at time t after injection will be predicted by a biexponential equation as follows:
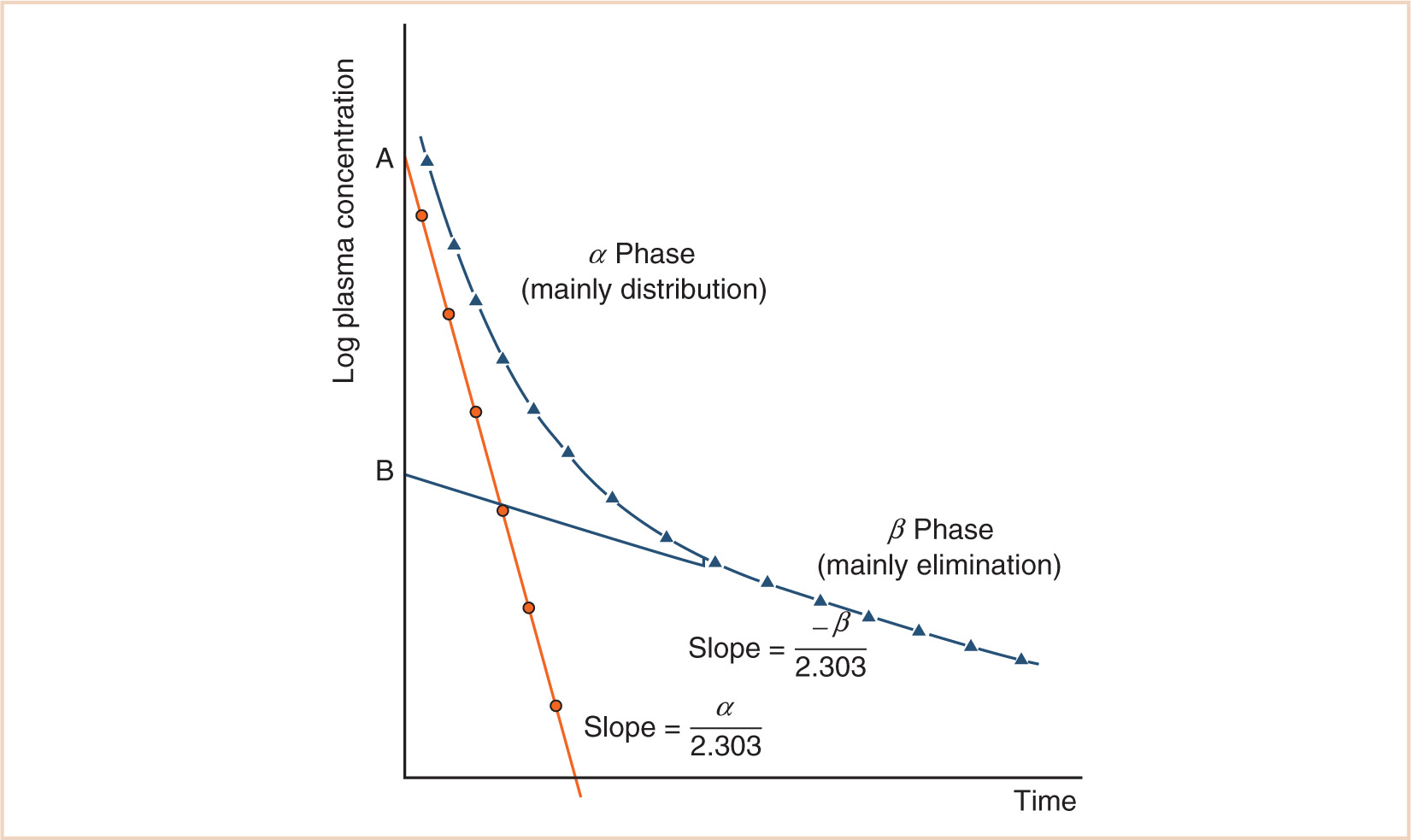
![]()
Each phase of the concentration–time curve is characterized by a constant (A or B), corresponding to a zero-time intercept on the Y-axis, and a hybrid rate constant (α or β) from which the corresponding half-times (T1/2α) and (T1/2β) may be calculated. The volume of the central compartment (V1) is calculated by dividing the total dose by A + B. The volume of distribution at steady state (Vdss = V1 + V2) is obtained by dividing the total dose by B.
These parameters (A, B, α, β) have little connection with underlying physiology, and an alternative parameterization is to use a central volume and three rate constants (k10, k12, k21) that describe drug distribution between compartments. Another common method is to use two volumes (central, V1; peripheral, V2) and two clearances (CL, Q); Q is the intercompartment clearance, and the volume of distribution at steady state (Vdss) is the sum of V1 and V2. Computers have made nonlinear regression techniques to directly estimate parameters easier through iterative techniques using least squares curve fitting. Models with two or more compartments are now commonly solved using differential equations rather than graphical techniques, for example, for a two-compartment mammillary model comprising a central compartment with volume V1 and concentration C1 and a peripheral compartment (V2, C2) with drug input (ratein)
![]()
![]()
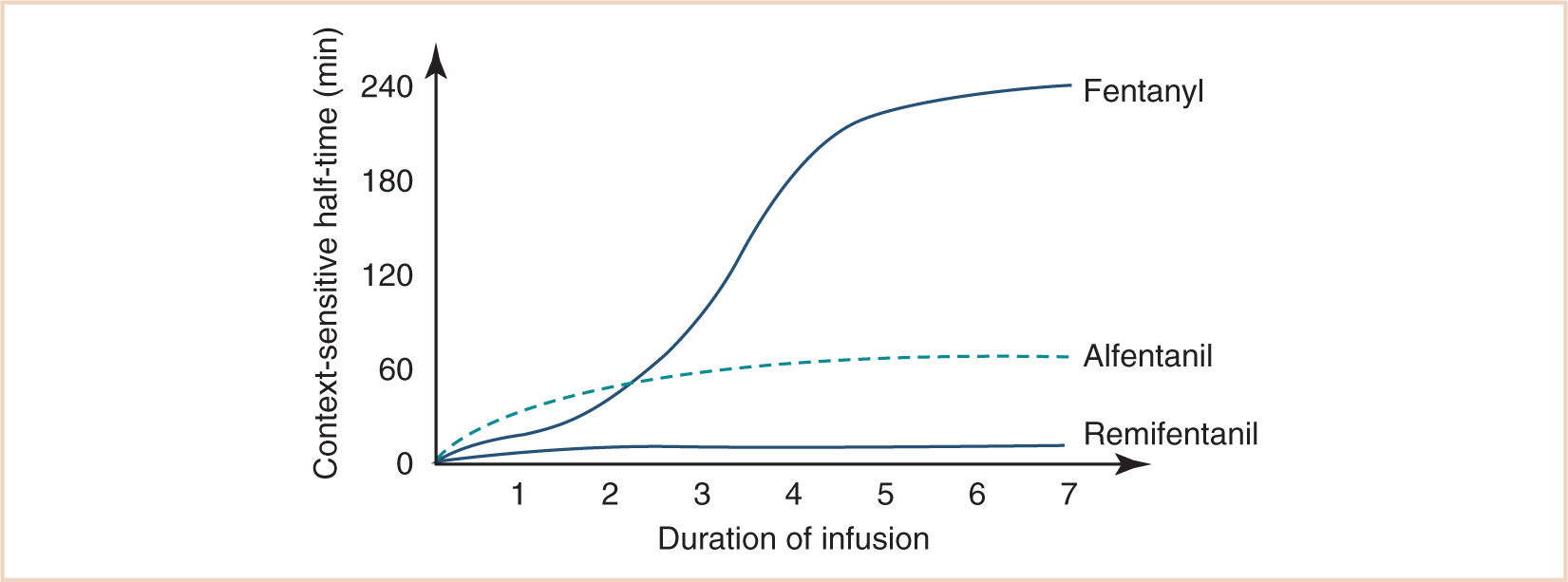
Most drugs used in anesthesia require two or more compartments to describe concentration changes with time, rendering half-life a poor parameter. Context-sensitive half-time refers to the half-time of a drug after an infusion of specified duration. For lipid-soluble drugs with a large Vdss, context-sensitive half-time will initially be short as the drug is redistributed to muscle and fat. As the infusion progresses, the deeper compartments fill up and the half-time lengthens until it equals the terminal half-time. A notable exception to this rule is remifentanil, whose context-sensitive half-time is fairly constant because its volume of distribution is small and its plasma clearance very rapid (see Fig. 2.2) (3).
FACTORS AFFECTING PHARMACOKINETICS IN INFANTS AND CHILDREN
The processes of absorption, distribution, and elimination of drugs are influenced by several age-related factors. In general, absorption and distribution of drugs tend to be increased in infants (children aged less than 12 months) compared with older subjects, while the capacity for elimination is often reduced. Thus, there is an increased risk of drug overdose and toxicity in the very young.
ABSORPTION
Absorption refers to the translocation of a drug from its site of administration into the systemic circulation. Most anesthetic drugs and adjuvants are administered by intravenous injection. Absorption by this route is rapid and complete. Absorption by other routes (e.g., intramuscular, inhalation) is often faster in infants, and this contributes to a more rapid onset of therapeutic as well as adverse effects. Enteral absorption when drugs are given orally is slower in neonates and infants. Gastric emptying and intestinal motor motility are delayed and may not mature until 6 to 8 months (4,5). This results in an increased time to peak concentration as well as decreased peak concentration compared to older children.
DISTRIBUTION
Distribution refers to the movement of a drug from the systemic circulation into the various body compartments.
Cardiac Output
At birth, weight-normalized resting cardiac output is around 200 mL/kg/minute, after which it declines gradually to about 100 mL/kg/minute by adolescence (see Fig. 2.3) (6). The higher cardiac output in infants and children translates into faster circulation times so that drugs are distributed to and from their sites of action more rapidly. As noted in Chapter 1, cardiac output varies with body weight3/4 since it is dependent on metabolic rate. However, when normalized for body weight as in Figure 2.3, cardiac output varies in relation to body weight−1/4 (i.e., wt3/4 ÷ wt1 = wt3/4−1 = wt–1/4). The same exponential decline will be seen for weight-normalized ECF volume (see Fig. 2.4) and the per kilogram doses of many anesthetic drugs in children aged over 1 year (7).
Protein Binding
Plasma protein binding limits the amount of drug that is free to diffuse into the extracellular space and interact with tissue receptors. In general, acidic drugs such as barbiturates bind mainly to albumin, while basic drugs such as the nonsteroidal inflammatory drugs and local anesthetics bind to globulins, lipoproteins, and glycoproteins. Several factors lead to decreased protein binding of drugs in the newborn period: (i) reduced concentrations of plasma proteins (see Fig. 2.5), (ii) the persistence of fetal albumin which has a reduced affinity for drugs, (iii) increased concentrations of free fatty acids and unconjugated bilirubin which compete with acidic drugs for binding sites, and (iv) a tendency to acidosis, which may alter the ionization and binding properties of plasma proteins and of drugs. Total plasma protein concentration and binding capacity approach adult values by about 1 year of age (8).
Body Water
Total body water constitutes 80% of the body weight at birth, but this declines dramatically in the first year to about 60% (see Fig. 2.4) (9). Most of the reduction in total body water is accounted for by a decrease in ECF, which declines from 45% of the body weight at birth to 26% by the age of 1 year. ECF volume declines further during childhood so that in the adult it constitutes only about 19% of the body weight. The greater volume of ECF in children is important because it constitutes part of the volume of distribution of all drugs. It is also a major thoroughfare for all nutrients and metabolites in the body, and therefore, it is perhaps not surprising to find that ECF volume varies with body weight3/4 (or body weight−1/4 when expressed as a percentage of body weight), suggesting a relationship to metabolic rate. Polar drugs such as aminoglycosides and NMBDs distribute rapidly into the ECF, but enter cells more slowly. The initial dose of such drugs is usually greater in infants and children compared with adults (e.g. succinylcholine) because of an increased ECF volume.
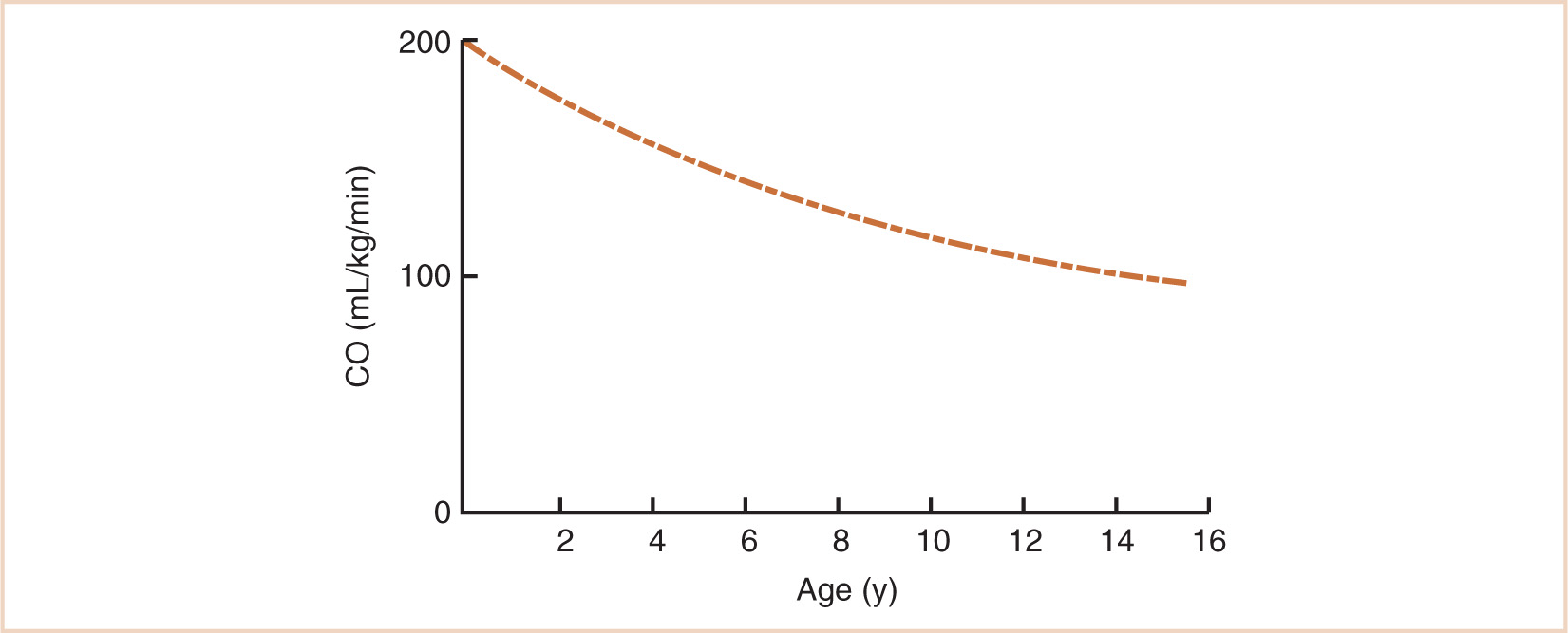
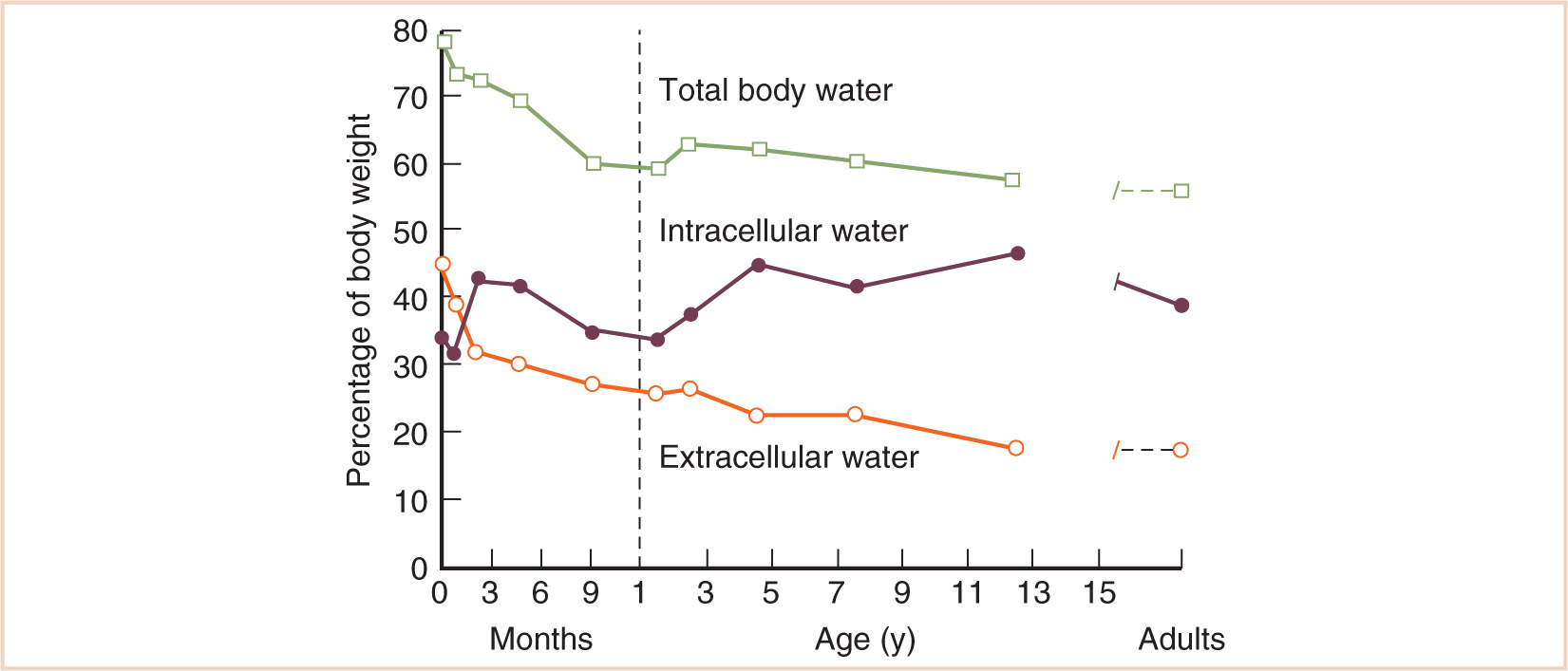
FIGURE 2.4 Changes in body water compartments with age.
(From Friis-Hansen B. Body water compartments in children: changes during growth and related changes in body composition. Pediatrics 1961; 28: 169–181)
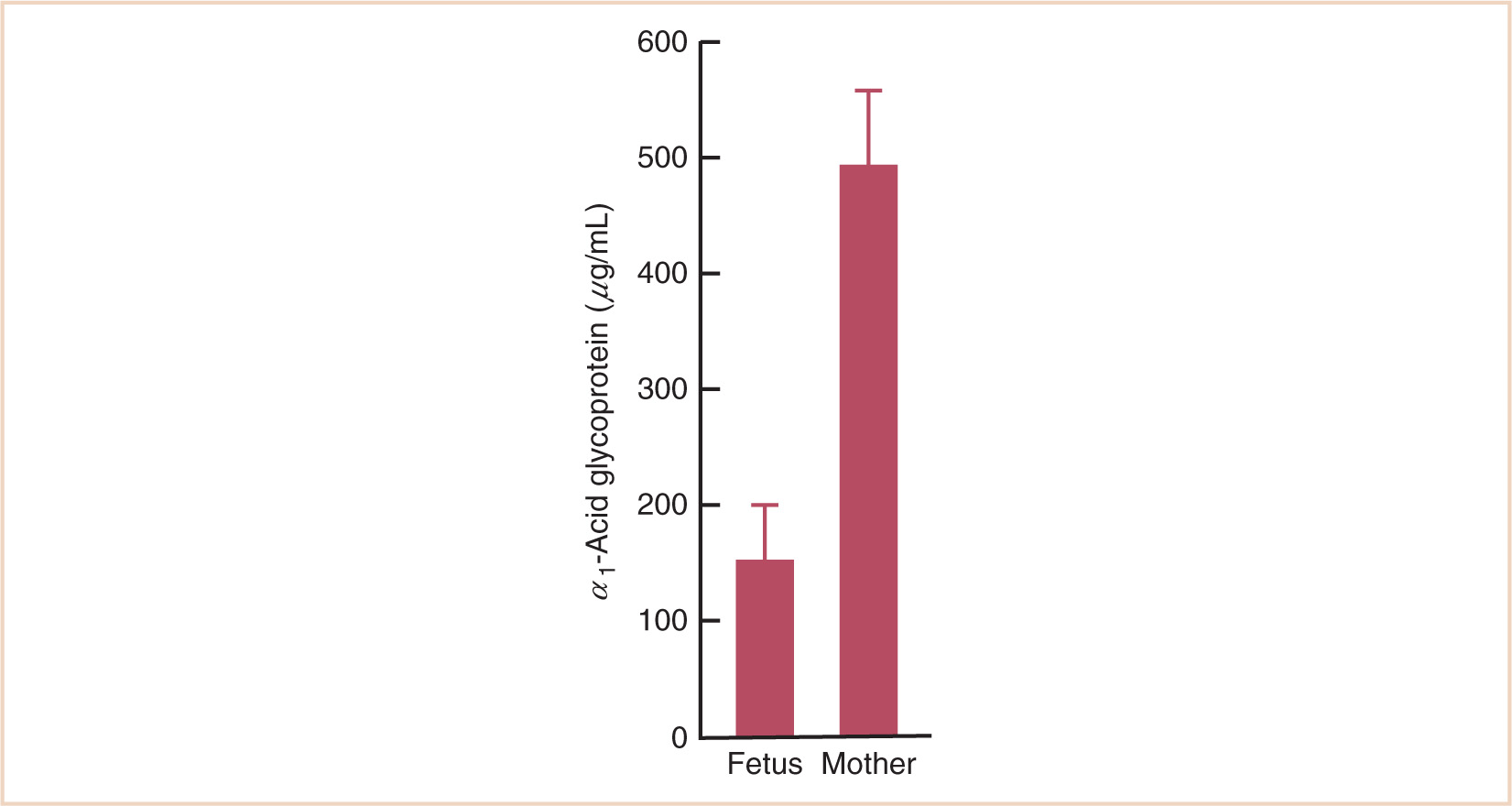
FIGURE 2.5 Concentration of α1-acid glycoprotein in the fetus and the mother. The concentration in the fetus is about one third of that in the mother.
The Blood–Brain Barrier
This term refers to the relative impermeability of brain capillaries to most ionized substances and macromolecules. The basis of this impermeability appears to be the presence of tight junctions between the endothelial cells of brain capillaries, and the absence of pinocytotic vesicles in brain capillary cytoplasm (10). Neonates are sensitive to most of the sedatives, opioids, and hypnotics, and this may in part be related to increased brain permeability (immature blood–brain barrier or damage to the blood–brain barrier) for some medications. There are also specific transport systems that mediate active transport. Pathologic CNS conditions can cause blood–brain barrier breakdown and alter these transport systems. Fentanyl is actively transported across the blood brain barrier by a saturable adenosine triphosphate-dependent process, while adenosine triphosphate-binding cassette proteins such as P-glycoprotein actively pump out opioids such as fentanyl and morphine. P-Glycoprotein modulation significantly influences opioid brain distribution and onset time, as well as the magnitude and duration of analgesic response (11).
ELIMINATION
Elimination refers to the removal of a drug from the body. It encompasses metabolism (biotransformation) and excretion.
Metabolism
Metabolic processes generally transform lipid-soluble drugs into water-soluble metabolites, which can be excreted more readily by the kidney. The reactions involved are usually categorized as phase I (nonsynthetic) or phase II (synthetic) reactions. Phase I reactions include oxidation, reduction, and hydrolysis, while phase II processes involve conjugation with other molecules, notably glucuronide, glycine, and sulfate. Most of the enzymes responsible for these reactions are found in the smooth endoplasmic reticulum of hepatocytes and are recovered from the microsomal fraction of cell homogenates.
Microsomal enzyme activity has been classified into three groups: those mature at birth, but decrease with age (e.g., CYP3A7 responsible for methadone clearance in neonates), those mature at birth and are sustained through adulthood (e.g., plasma esterases that clear remifentanil (12)), and those that are immature at birth (13). These latter are the majority. In the newborn, the concentrations and activities of many microsomal enzymes are reduced or absent. In particular, parts of the mixed oxidase or cytochrome P450 system are less than half as active as in adults, and drug clearance may take from a few weeks to several years to develop (14,15). This results in reduced metabolism of most drugs. Similarly, the ability to form glucuronide conjugates is depressed due to a reduction in the activity of glucuronyl transferase and uridine 5′-diphospho-glucose (UDPG) dehydrogenase. A clinical consequence of this is the unconjugated bilirubinemia commonly observed in premature babies. This is of major significance in the clearance of drugs such as morphine, acetaminophen, and dexmedetomidine, which is greatly depressed in the neonate and takes over 6 months to approach adult rates (see Fig. 2.6) (16). The clearance of these drugs by individual isoforms of the UDPG dehydrogenase system mirrors maturation of renal function.
CLINICAL PEARL Some drugs (e.g., atracurium, succinylcholine, remifentanil) are metabolized at extrahepatic sites, often by esterases. Plasma clearance is independent of organ function and, when normalized for body weight, is somewhat greater in neonates and infants than in older children. Remifentanil clearance determines infusion rate at steady state, and remifentanil infusion rates required to achieve a target concentration are greater in neonates and infants.
Many drugs are metabolized at extrahepatic sites, often by esterases. Atracurium is metabolized by nonenzymatic cleavage at body temperature and pH (Hofmann elimination) as well as by hydrolysis. Thus, plasma clearances of atracurium and remifentanil are independent of organ function and, when normalized for body weight, are somewhat greater in neonates and infants than in older children (17,18). This relationship is demonstrated for remifentanil in Figure 2.7. Clearance determines infusion rate at steady state, and it can be seen that remifentanil infusion rates required achieving a target respiratory rate mirror clearance. Succinylcholine also exhibits a monophasic decline in weight-normalized dose requirements from birth, which is predictable by the 3/4 power size model (19). This latter observation suggests that the postjunctional elements of neuromuscular transmission are well developed at birth.
Genetic influences may also have a profound effect on metabolic clearance pathways (20,21). Reduced succinylcholine clearance by butyrylcholinesterase and reduced isoniazid acetylation activity are well-known examples. Single nucleotide changes or polymorphisms (SNPs) in the DNA sequence in CYP enzymes usually decrease but may also increase metabolic activity for a specific drug or drug substrate (22). The CYP2D6 drug-metabolizing enzyme is responsible for clearance of a number of drugs, including amitriptyline, codeine, tramadol, and hydrocodone (23). For the 2% to 10% of the population who are CYP2D6 poor metabolizers, codeine causes limited opioid effects. However, individuals with duplicated active CYP2D6 genes are classified as ultraextensive metabolizers, and the rapid ultrarapid metabolism of codeine into morphine can result in narcosis, apnea and death (24).
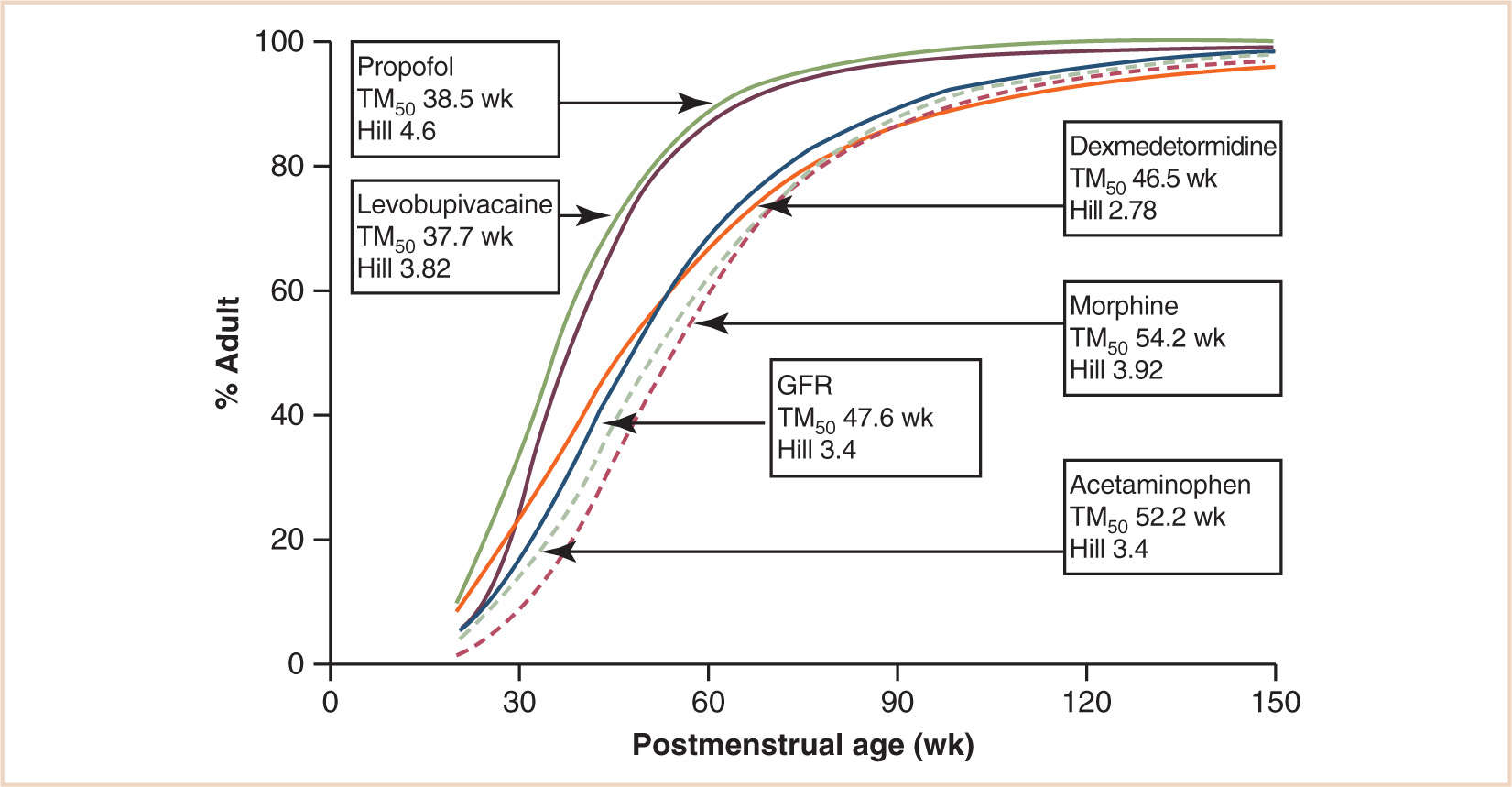
FIGURE 2.6 Clearance maturation, expressed as a percentage of mature clearance, of drugs where glucuronide conjugation (paracetamol, morphine, dexmedetomidine) plays a major role. These profiles are closely aligned with GFR. In contrast, cytochrome P450 isoenzymes also contribute to propofol and levobupivacaine metabolism and cause a faster maturation profile than expected from glucuronide conjugation alone.
(From Anderson BJ, Holford NH. Tips and traps analyzing pediatric PK data. Paediatr Anaesth. 2011;21:222–237, with permission.)
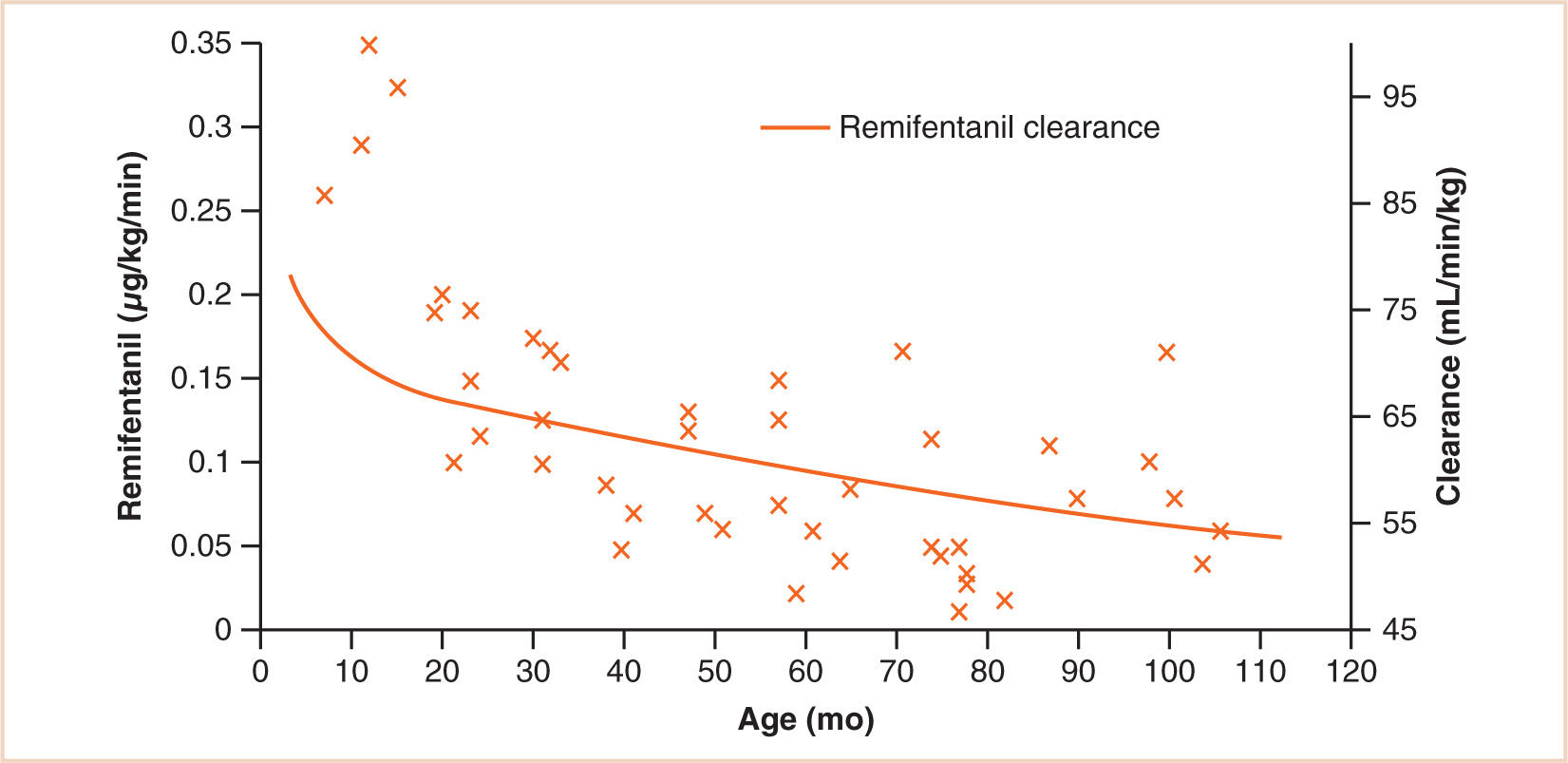
FIGURE 2.7 The effect of age on the dose of remifentanil tolerated during spontaneous ventilation (respiratory rate 10 breaths per min) during anesthesia in children undergoing strabismus surgery. Superimposed on this plot is remifentanil clearance. There is a mismatch between clearance and infusion rate for those individuals still in infancy where we might anticipate a respiratory rate greater than 10 breaths per min.
(From Anderson BJ. Pediatric models for adult target-controlled infusion pumps. Paediatr Anaesth. 2010;20:223–232, with permission.)
Excretion
Drugs and their metabolites are mainly excreted by the kidney; the processes involved are glomerular filtration and tubular secretion. Some drugs are simply filtered, in which case their rate of elimination will depend upon the glomerular filtration rate (GFR). GFR is lower in neonates and infants than in adults, and when related to body surface area (BSA), it attains adult values at 3 to 5 months of age (25). A similar maturation profile has been demonstrated using an allometric ¾ power model with postmenstrual age (PMA) as the ordinate. Glomerular maturation is progressive before birth and reaches half adult rates at 48 weeks PMA (see Fig. 2.6) (26). Proximal tubular secretion, which is important for the elimination of some conjugated drugs, reaches adult rates by about 7 months of age (25).
FACTORS AFFECTING PHARMACODYNAMICS DURING DEVELOPMENT
While pediatric pharmacokinetic changes with age are increasingly reported, there remains a dearth of literature concerning pharmacodynamic changes (27). The interaction between a drug and its receptor may be influenced by developmental changes in receptor number, type, affinity, and the availability of natural ligands. In addition, organ size and blood flow changes that occur with age impact on drug concentration at the effect site. Such changes occur commonly in the newborn period and have been implicated in the altered responses to some anesthetic drugs. Common examples include the inotropic effect of calcium on the neonatal myocardium and the limited effect of salbutamol on neonatal bronchial muscle.
NICOTINIC ACETYLCHOLINE RECEPTOR
The nicotinic acetylcholine receptor (AChR) is a ligand-gated ion channel with a molecular weight of 260 kilodaltons (kDa), which in mammals exists in fetal and adult forms (28,29). The fetal form is similar to the AChR receptor of the electric eel and consists of five protein subunits designated α, α, β, γ, and δ grouped around a central pore (see Fig. 2.8). In the adult form, the γ subunit is replaced by a subunit designated ε, which decreases the open time of the receptor after two molecules of acetylcholine bind to its α subunits. The longer open time of the fetal receptor increases the safety factor in neuromuscular transmission and may explain the observation that newborn rats are resistant to tubocurarine (30). The fact that resistance to tubocurarine is not normally seen in human newborns is consistent with histological evidence that fetal AChRs are not present on human muscle fibers after 31 weeks of gestational age (31).
Combined pharmacodynamic and pharmacokinetic studies suggest that the neuromuscular junction of the human neonate is three times more sensitive to the effects of nondepolarizing muscle relaxants than that of the adult (32,33). A similar degree of sensitivity has been demonstrated in 11-day-old rats and shown to be due to a 3-fold reduction in the release of acetylcholine from the motor nerve (see Fig. 2.9) (30,34).
OPIOID RECEPTORS
Radioreceptor binding studies in the rat have shown that the relative proportion of μ, κ, and δ opioid receptors is age dependent (35). Furthermore, work in mice has shown that the selectivity of the μ receptor for μ-specific ligands changes with age (36). As the µ and κ receptors are known to mediate the respiratory effects of opioids, changes in the number and affinity of these receptors could be a factor in the increased susceptibility of newborns to opioid-induced respiratory depression.
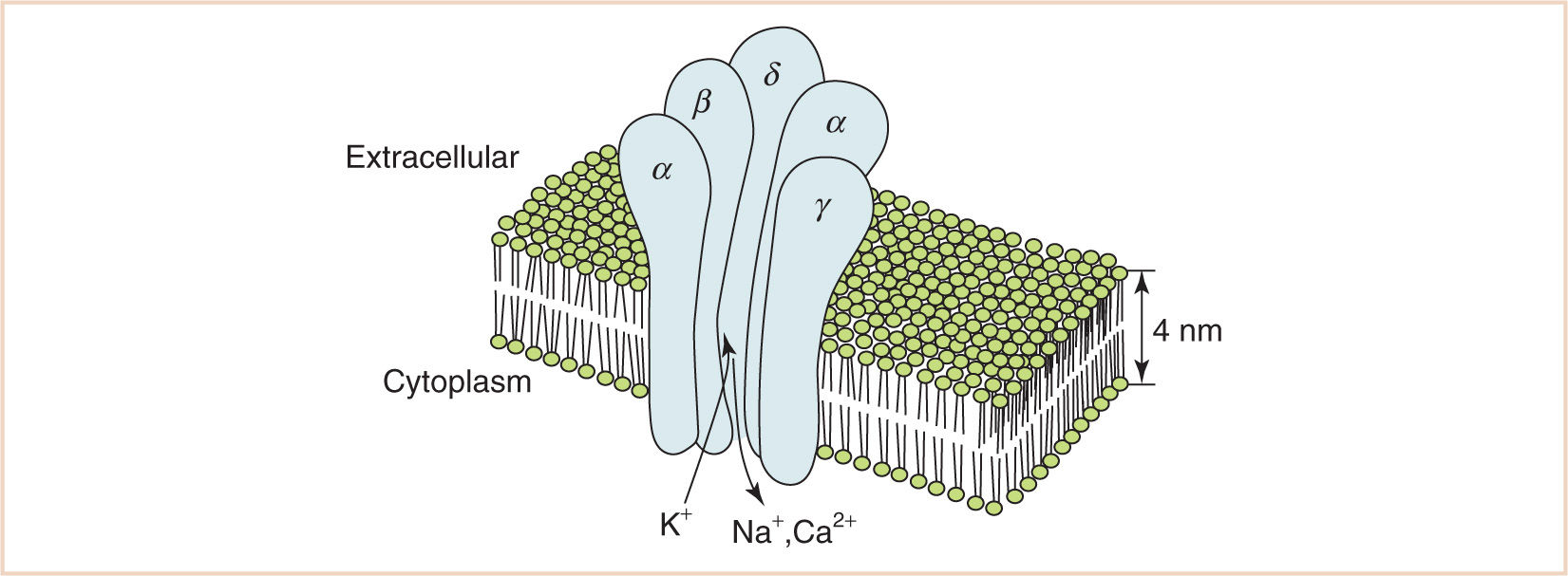
FIGURE 2.8 Fetal AChR. Five glycoprotein subunits designated α, α, β, γ, and δ are grouped around a central pore. In the adult AChR, subunit γ is replaced by a subunit designated ε.
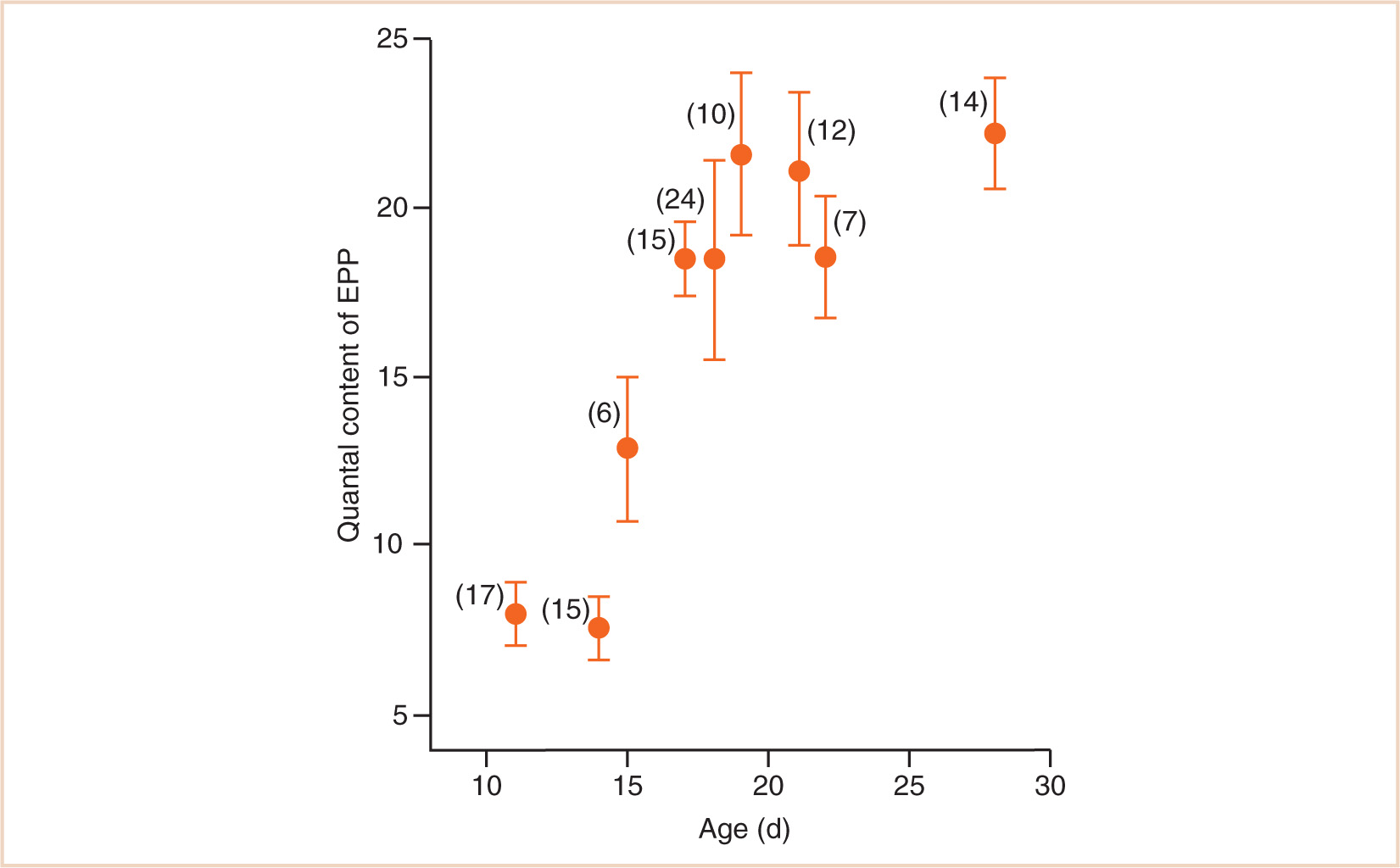
FIGURE 2.9 Variation in quantal content of the endplate potential phrenic nerve-hemidiaphragm preparations from rats aged 11 to 28 days. The 3-fold reduction in ACh release at 11 days compared to 21 days corresponds with a 3-fold increase in sensitivity of the neuromuscular junction to tubocurarine.
δ-AMINOBUTYRIC ACID AND N-METHYL-D-ASPARTATE RECEPTORS
The δ-aminobutyric acid (GABAA) and N-methyl-D-aspartate (NMDA) receptors are ligand-gated ion channels with features similar to those of the AChR. The GABAA receptor is the site of action of benzodiazepines and barbiturates, and a likely target for most general anesthetics (37). The NMDA receptor regulates L-glutamate signaling and is blocked by the action of ketamine. Both receptors undergo profound developmental changes in their subunits and roles, which may have important implications for anesthesia and normal neuronal development (38–41).
PEDIATRIC DOSING
Deciding the correct dose of a drug for an infant or child invariably involves scaling the dose according to the patient’s size (42). However, it is important to appreciate that such scaling does not take account of age-related drug sensitivities and drug handling. Moreover, different doses will be obtained depending on which of several available size models is used (43).
SCALING FOR SIZE
The basic principles of scaling for size or allometry (Gk. Alo-other, metron–measure) have been explored in Chapter 1. One of the first attempts at scaling for size with anything other than body weight was the surface area law, which asserts that physiological functions are comparable whatever the size of the organism when related to BSA. It was subsequently shown that BSA varies with body weight to the power of two thirds, but that the best approximation for metabolic processes is obtained by using an exponent of 3/4 as shown in Figure 1.1. As a direct extension of this work, the size models most commonly used to scale maintenance pediatric doses are the weight model, the 2/3 power model (surface area model), and the 3/4 power model. The following equations illustrate how these size models may be used to obtain pediatric doses from adult ones.
1. Weight model | |
| Dosepediatr = (Wpediatr/Wadult) × Doseadult |
2. 2/3 power (surface area) model | |
| Dosepediatr = (Wpediatr/Wadult)2/3 × Doseadult |
3. 3/4 power model | |
| Dosepediatr = (Wpediatr/Wadult)3/4 × Doseadult |
CLINICAL PEARL Maintenance dose (Dosepediatr) in a child (greater than 1 year) can be estimated from adult dose using the weight-based (W) formula
Dosepediatr = (Wpediatr/Wadult)3/4 × Doseadult
These models are only applicable out of infancy, after maturation of clearance pathways (44). An additional function describing maturation is required for neonates and infants (see Fig. 2.10). Figure 2.11 compares these three size models. The per kilogram model underpredicts dose in children. The surface area model predictions are close to those predicted from the allometric ¾ model, although the BSA model overpredicts in infants. The strength of the allometric model is that clearance prediction is possible over a size range of 14 orders of magnitude (see Fig. 1.2) (45). Propofol clearance in children has been predicted from that in rats using allometry (46).
These formulae are not applicable for calculation of an initial dose because that is dependent on volume of distribution.
Initial dose = target concentration × volume of distribution
Maintenance dose = target concentration × clearance
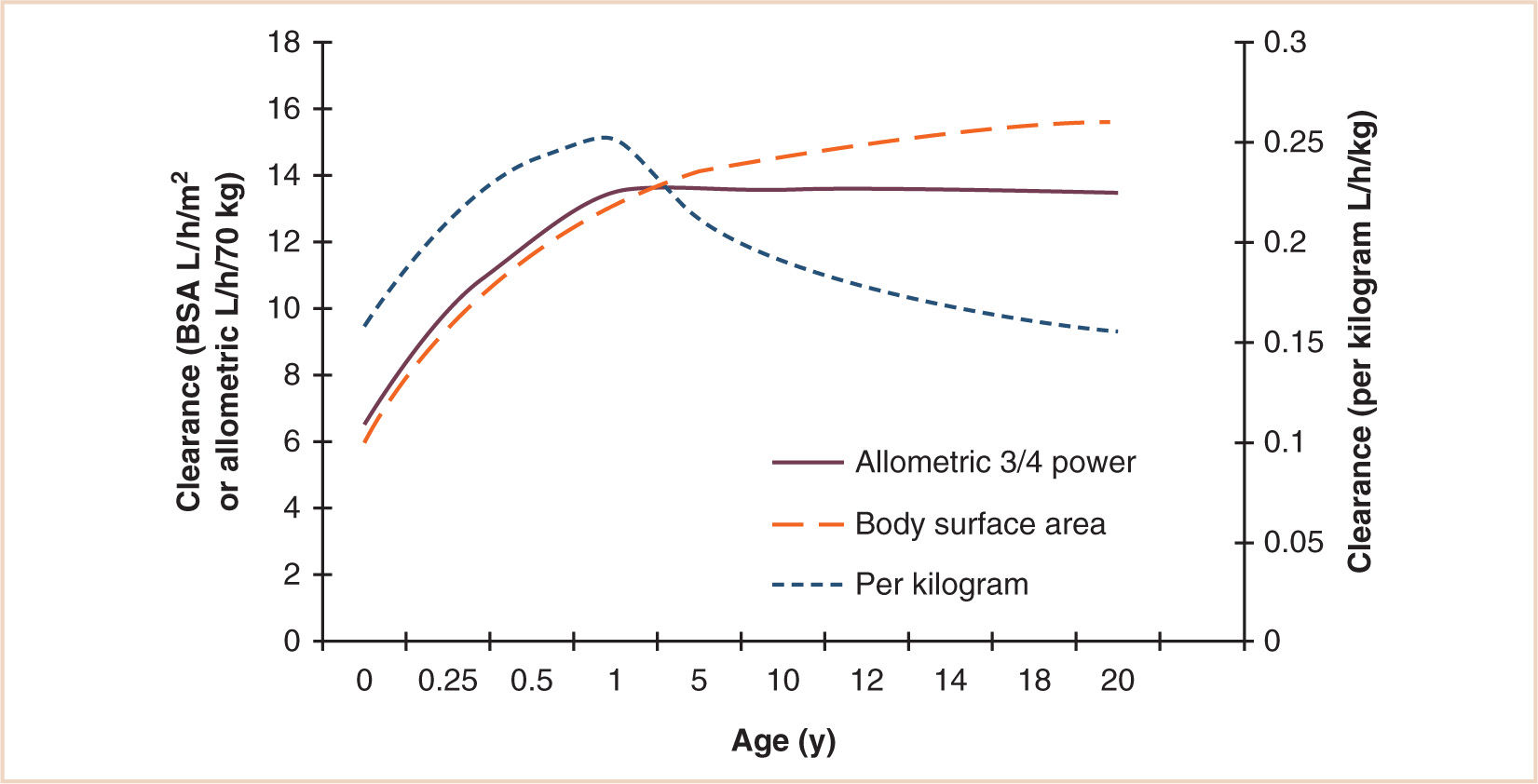
FIGURE 2.10 Age-related clearance changes for a hypothetical drug. All three models show an increase in clearance over the first year of life due to maturation of metabolic pathways. Clearance expressed using the per kilogram model then decreases with age after 1 year to reach adult rates in adolescence. This course is not evident with the allometric ¾ power and surface area models.
(From Anderson BJ, Meakin GH. Scaling for size: some implications for paediatric anaesthesia dosing. Paediatr Anaesth. 2002;12:205–219, with permission.)
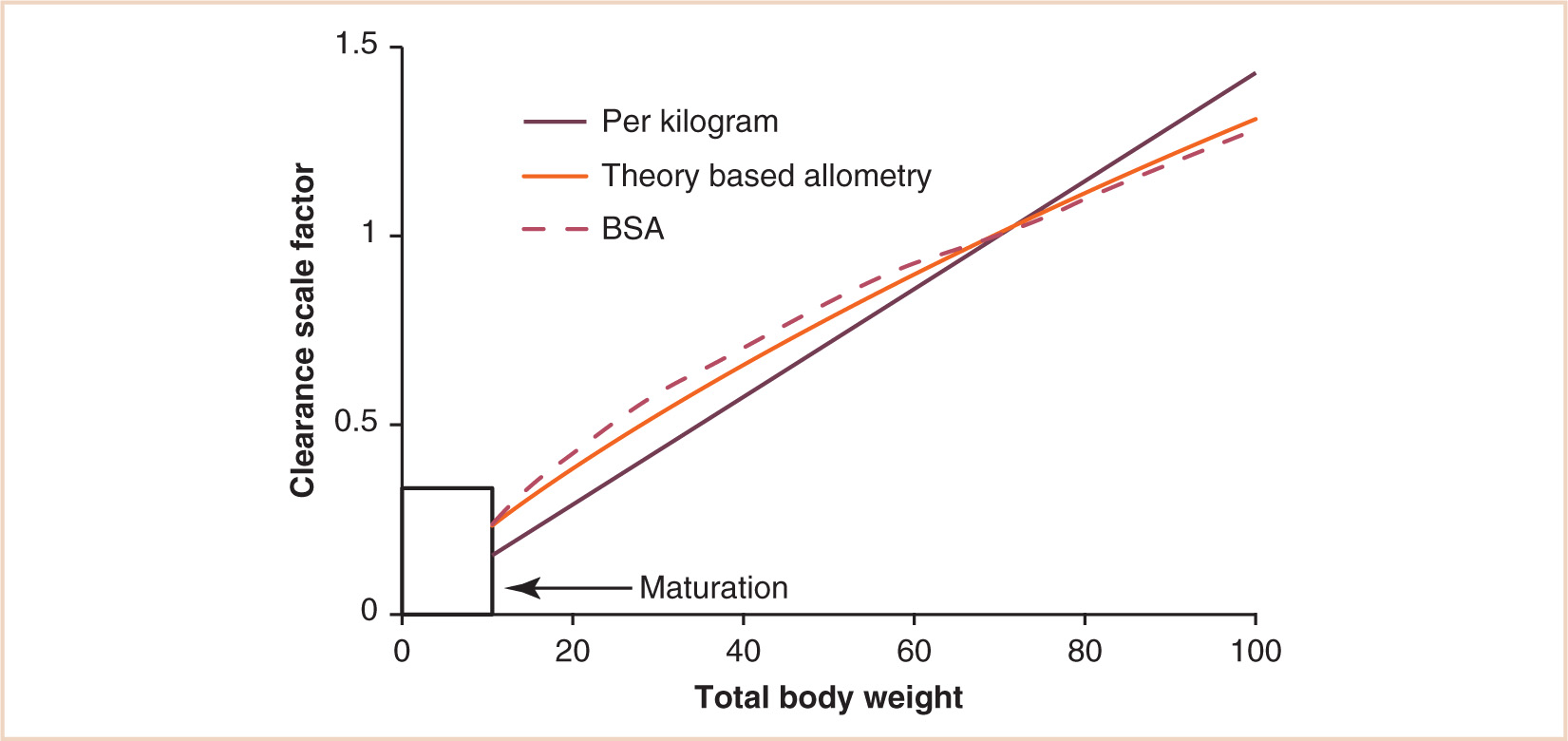
FIGURE 2.11 Body size metrics used to describe clearance changes with weight. A nonlinear relationship exists between weight and clearance. The linear model underestimates clearance in children, while the BSA model overestimates clearance in children.
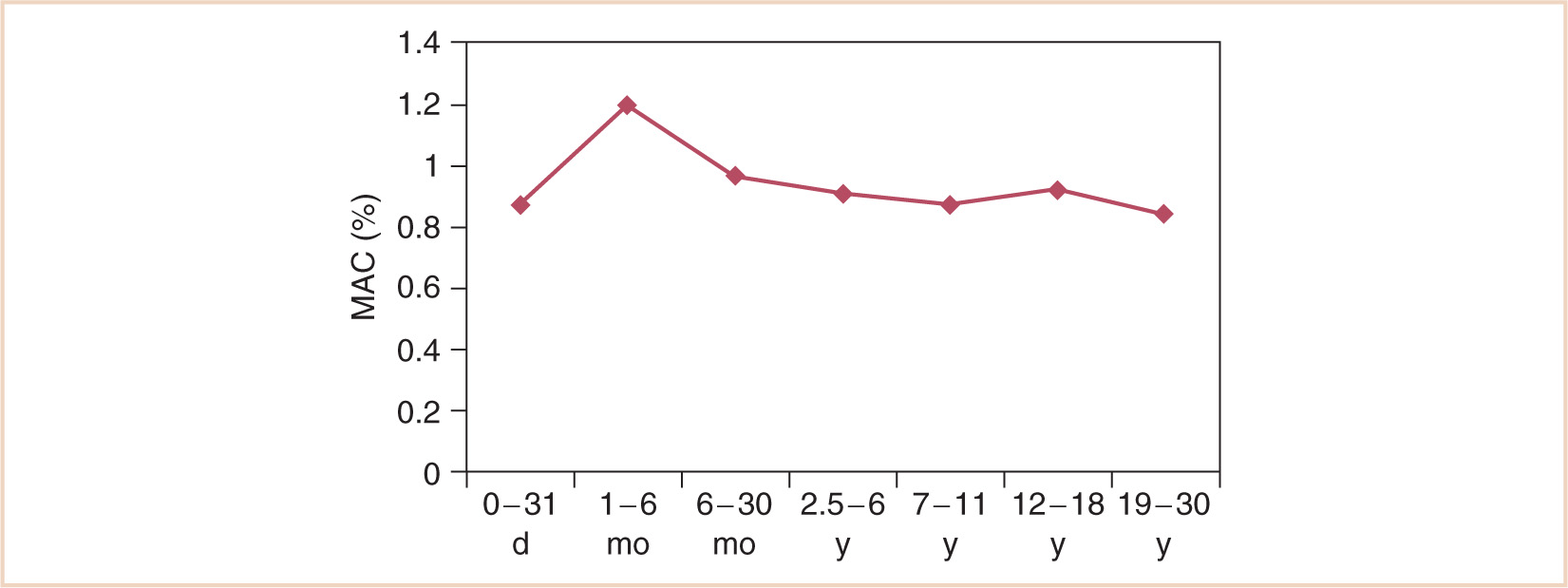
FIGURE 2.12 Changes in the MAC of halothane with age.
(Adapted from Lerman J, Robinson S, Willis MM, et al. Anesthetic requirements for halothane in young children 0–1 month and 1–6 months of age. Anesthesiology. 1983;59:421–423; Gregory G, Eger EI, Munson ES. The relationship between age and halothane requirements in man. Anesthesiology. 1969;30:488–491.)
The biphasic distribution of drug dosage shown in Figure 2.10 is shared by many drugs, including most volatile anesthetics (see Fig. 2.12) (47) and the nondepolarizing muscle relaxants (see Fig. 2.13) (48), although the mechanisms contributing to these changes comprise complex interactions between pharmacokinetics and pharmacodynamics. The ascending limb reflects, in part, age-dependent maturation of organ or enzyme systems that are not predictable by size models. This is true of Figure 2.10, which simply shows clearance changes with age. However, Figure 2.13 demonstrates response changes with age after a single dose. Consequently, volume of distribution changes as well as receptor numbers and structural changes with age also contribute to response. The neuromuscular junction, for example, is immature in neonates, while muscle bulk (and receptor numbers) dramatically increase during childhood. Reduced clearance in neonates prolongs duration of action. Cerebral blood flow, solubility, GABAA receptor numbers, or developmental shifts in the regulation of chloride transporters may also contribute to volatile anesthetic changing requirements with age (see Fig. 2.12).
In summary, it seems likely that the best estimate of maintenance dosing in a child from that of an adult will be obtained using the ¾ power model, which relates drug doses to metabolic rate. However, none of the size models is 100% reliable, and their use must be supported by pharmacodynamic and pharmacokinetic data. Doses are much less predictable in infants aged less than 1 year due to immaturity of organs and enzyme systems. Reduced drug doses are frequently required in this age group.
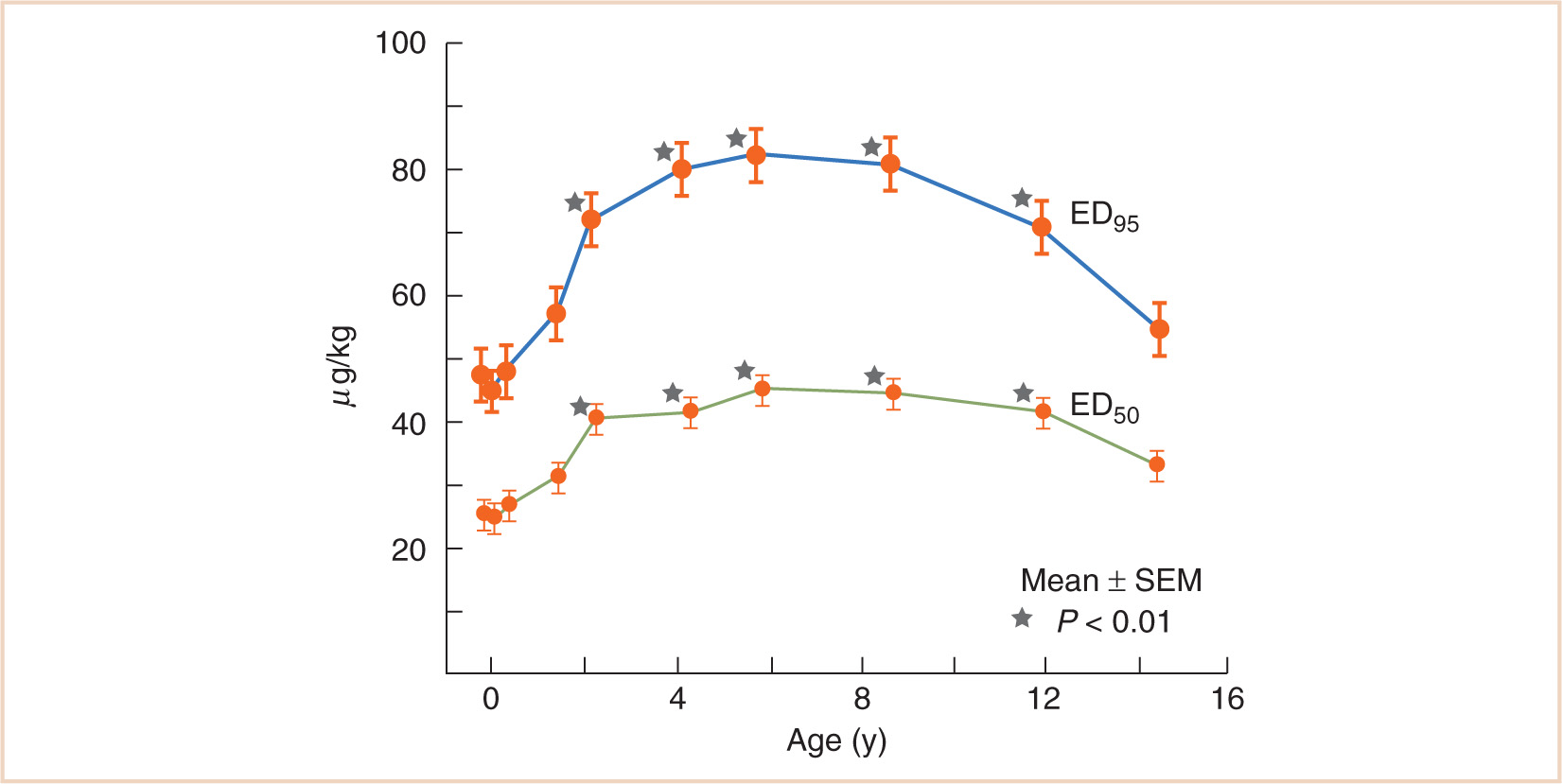
FIGURE 2.13 Variation in the effective doses of vecuronium for 50% and 95% depression of twitch (ED50, ED95) with age. The values are significantly greater in children aged 2–13 years compared with younger and older patients.
(Adapted from Meretoja OA, Wirtavuori K, Neuvonen PJ. Age-dependence of the dose-response curve of vecuronium in pediatric patients during balanced anesthesia. Anesth Analg. 1988;67:21–26.)
INTRAVENOUS ANESTHETIC AGENTS
Intravenous anesthetics are a heterogenous group of sedative-hypnotic drugs that produce unconsciousness in a single arm-brain circulation time when injected intravenously. Prompt awakening after a single dose of these agents occurs predominantly by redistribution.
THIOPENTAL
Thiopental is an analog of pentobarbital, in which the oxygen attached at C2 of the barbituric acid ring is replaced by sulfur. This substitution confers high lipid solubility, which in conjunction with a high cerebral blood flow, results in rapid penetration of the brain and hypnosis. Elimination occurs by oxidation in the liver to an inactive metabolite which is excreted by the kidney.
In the neonate, plasma protein binding of thiopental is reduced so that the fraction of unbound drug is almost twice that found in older children and adults (49,50). In addition, clearance at 26 weeks PMA was 0.015 L/minute/70 kg and increased to 0.119 L/minute/70 kg by 42 weeks PMA (~ 40% of adult clearance at term (51); however, as recovery depends mainly on redistribution, the effect of an induction dose is not significantly prolonged (see Fig. 2.14).
Children aged 13 to 68 months given rectal thiopental (44 mg/kg) 45 minute prior to surgery were either asleep or adequately sedated with plasma concentrations above 2.8 mg per L (52). The ED50 sleep dose of intravenous thiopental varies with age (53,54). Doses of about 1.3 × ED50 of thiopental are required to produce rapid, reliable induction of anesthesia in all age groups; thus, healthy neonates require about 4 to 5 mg per kg, infants 7 to 8 mg per kg and children 5 to 6 mg per kg of thiopental for induction. The reduced requirement for thiopental in neonates compared with infants aged 1 to 6 months may be explained by decreased plasma protein binding (49), greater penetration of the neonatal brain (55), or increased responsiveness of neonatal receptors (56). The increased requirements for thiopental in infants and children compared with adults (average adult dose 4 mg/kg) remain unknown. A pharmacodynamic explanation attributable to increased cerebral GABAA receptor numbers or maturational differences in relative organ mass and regional blood flow may contribute. Blood flow, relative to cardiac output, to kidney and brain increases, while that to the liver decreases early life. Cerebral and hepatic mass as a proportion of body weight are much higher in the young child than in the adult.
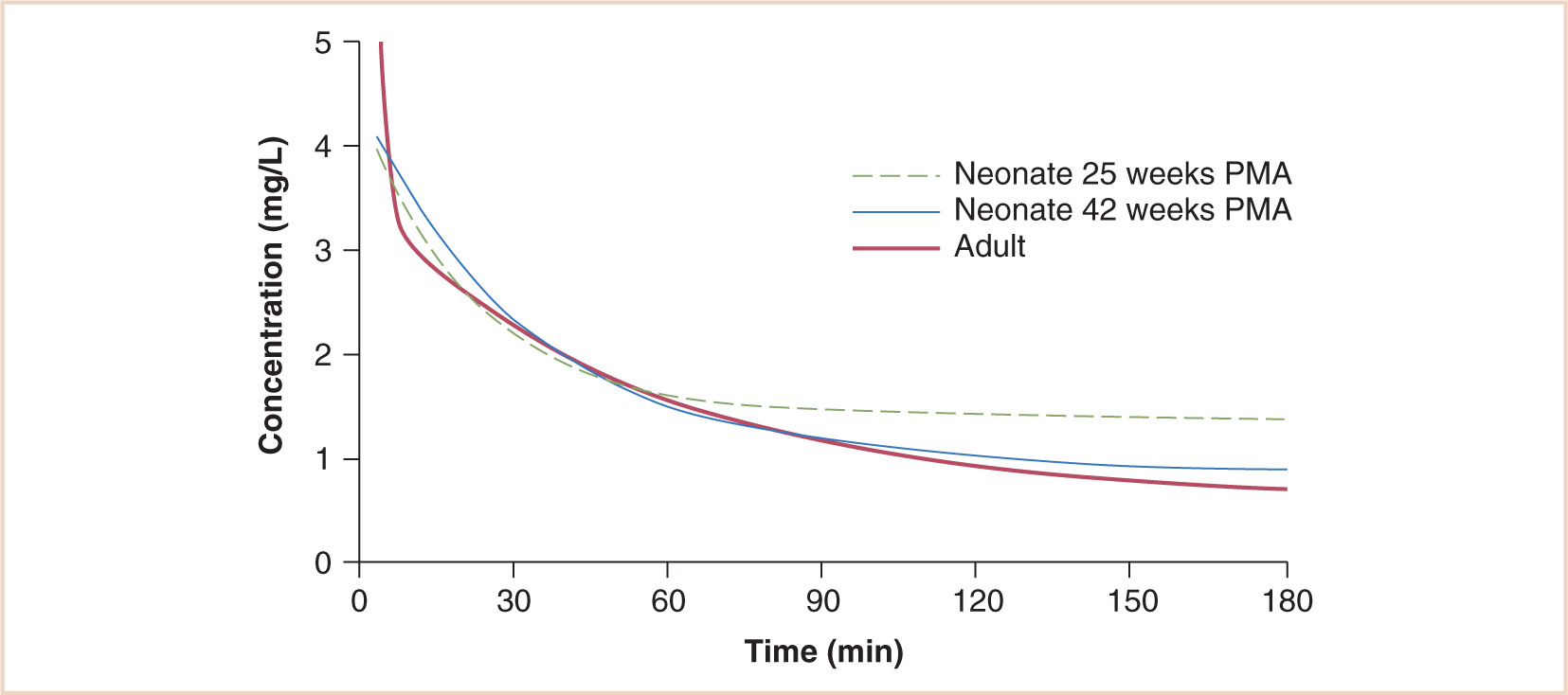
FIGURE 2.14 Simulated time–concentration profiles after an intravenous thiopentone bolus of 3 mg per kg. Predictions for a 25-week PMA and a 42-week PMA neonate are shown alongside that of an adult.
(From Larsson P, Anderson BJ, Norman E, et al. Thiopentone elimination in newborn infants: exploring Michaelis-Menten kinetics. Acta Anaesthesiol Scand. 2011;55:444–451, with permission.)
A few seconds of apnea followed by a period of respiratory depression is common after an induction dose of thiopental. The hypotensive response in neonates given thiopental appears not as dramatic as that associated with propofol although it still may occur with reversion to fetal circulation. Thiopental has little direct effect on vascular smooth muscle tone. In two studies, induction of anesthesia with thiopental was accompanied by a 14% to 23% reduction in arterial blood pressure in healthy infants and children, secondary to myocardial depression and a reduction in peripheral vascular resistance (57,58); several other studies found no changes in arterial pressure after giving thiopental to these age groups (54,59,60). The degree of cardiovascular depression after administration of thiopental may be related to the dose given and the rate of injection. Thiopentone is a direct myocardial depressant and may cause significant systemic hypotension in the hypovolemic state. Other reported adverse effects include hiccups, coughing, and laryngospasm. Extravasation of thiopental or intra-arterial injection can cause tissue injury, probably due to its extreme alkalinity.
Thiopental has also been used as a continuous infusion (2 to 4 mg/kg/hour) to control intracranial hypertension. The elimination of thiopental after a continuous infusion may be markedly prolonged compared with that after a single bolus (11.7 vs. 6.1 hours) (61). These findings may in part be attributed to the underlying illness, intercurrent drug treatment, and zero-order kinetics at higher concentrations (Michaelis constant 28.3 mg/L) (51,62).
PROPOFOL
Propofol is an isopropylphenol supplied as a 1%, 2%, or 10% aqueous solution containing soybean oil, glycerol, and purified egg phosphatide to improve solubility. Its use for induction and maintenance of anesthesia is associated with rapid recovery. Although the package insert for propofol cautions against its use in all patients with “egg allergy,” the evidence for this is not convincing (63,64). Propofol also suppresses laryngeal and pharyngeal reflexes, thereby facilitating tracheal intubation and the insertion of a laryngeal mask airway (65–67). Emergence delirium rarely occurs after propofol anesthesia in children. Propofol reduces the incidence of nausea and vomiting when used as an induction agent or when used for the maintenance of anesthesia (68). In view of these advantages, propofol has replaced thiopental as the induction agent of choice.
CLINICAL PEARL Emergence delirium is increased in children aged less than 5 years. Prevention of emergence delirium by using appropriate analgesia, more soluble volatile agents for maintenance, awareness of high-risk surgery, and using adjuncts such as fentanyl, α-2 agonists, ketamine, or propofol at the end of anesthesia should be considered.
TABLE 2.1 The pharmacokinetics of propofol and thiopental in children
| Propofol | Thiopental |
Vc (L/kg) | 0.6 | 0.4 |
Vdss (L/kg) | 5.0 | 2.1 |
Clearance (mL/kg/min) | 40.4 | 6.6 |
Distribution T1/2α (min) | 3.1 | 6.3 |
Elimination T1/2β (min) | 24.3 | 43.0 |
Terminal T1/2γ (h) | 3.5 | 6.1 |
Adapted from Jones RD, Chan K, Andrew LJ, et al. Pharmacokinetics of propofol in children. Br J Anaesth. 1990;65:661–667; Sorbo S, Hudson RJ, Loomis JC. The pharmacokinetics of thiopental in pediatric surgical patients. Anesthesiology. 1984;61:666–670.
TABLE 2.2 Pharmacokinetics of propofol in children and adults
| Children | Adults |
Vc (L/kg) | 0.6 | 0.4 |
Vdss (L/kg) | 5.0 | 11.7 |
Clearance (mL/kg/min) | 40.4 | 27.7 |
Distribution T1/2α (min) | 3.1 | 2.0 |
Elimination T1/2β (min) | 24.3 | 52.4 |
Terminal T1/2γ (h) | 3.5 | 11.2 |
Children’s data from Jones RD, Chan K, Andrew LJ, et al. Pharmacokinetics of propofol in children. Br J Anaesth. 1990;65:661–667; Adults’ data from Kirkpatrick T, Cockshott ID, Douglas EJ, et al. Pharmacokinetics of propofol (diprivan) in elderly patients. Br J Anaesth. 1988;60:146–150.
The pharmacokinetics of propofol and thiopental in children are compared in Table 2.1 (50,69). The shorter distribution half-time and more rapid plasma clearance of propofol are responsible for the faster and more clear-headed recovery following a single dose of this agent compared with thiopental (70). The rapid elimination of propofol (plasma clearance up to 10 times faster than thiopental in some adult studies) reduces the potential for accumulation, making the drug suitable for maintenance of anesthesia. Induction and maintenance doses of propofol are higher in children than in adults because the volume of the central compartment is 50% larger and the plasma clearance is 25% faster in children (Table 2.2) (69,71). Average induction doses (1.3 × ED50) in infants and children and adults are 4, 3, and 2 mg per kg, respectively (72,73). Clearance is limited by the hepatic blood flow and is consequently reduced in children in low cardiac output states.
Propofol is now commonly infused as part of a total intravenous anesthesia (TIVA) technique. Clearance is increased in children; consequently, a higher infusion dose is required to achieve the same target concentration as adults (e.g., 3 mg/L). A simple manual regimen for propofol infusion in adults (74) consists of a bolus of 1 mg per kg followed by an infusion of 10 mg/kg/hour (0 to 10 minutes), 8 mg/kg/hour (10 to 20 minutes), and 6 mg/kg/hour thereafter. Requirements for children, however, are greater. A loading dose of 2.5 mg per kg followed by an infusion rate of 15 mg/kg/hour for the first 15 minutes, 13 mg/kg/hour from 15 to 30 minutes, 11 mg/kg/hour from 30 to 60 minutes, 10 mg/kg/hour from 1 to 2 hours, and 9 mg/kg/hour from 2 to 4 hours resulted in a steady state target concentration of 3 mg per L in children aged 3 to 11 years (75). Increased requirements in children can be attributed to size factors.
Popular pediatric PK parameter sets used for propofol infusion targeting a plasma concentration are based on data from Marsh (76) and Gepts (77) (Diprifusor), Kataria (78), and Absalom (79) (Paedfusor) (Table 2.3). Parameter estimates are different for each author. Covariate influences such as severity of illness are unaccounted for; the volume in the central compartment, for example, is increased in children after cardiac surgery (80).
TABLE 2.3 Manual propofol infusion regimens to achieve a target concentration of 3 mg/L in adults and children 3–11 years
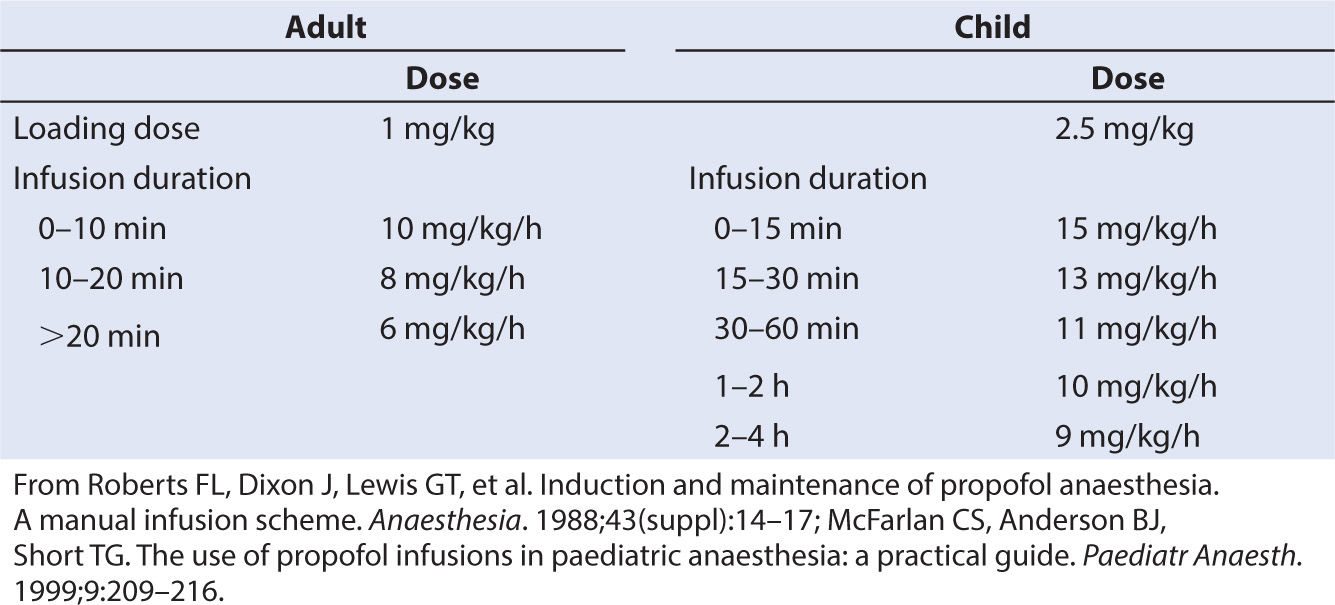
Decreased requirements in neonates are due to immature enzyme clearance systems (see Fig. 2.6). Propofol infusion rates for infants have been suggested (81). Those regimens were determined by adapting an adult dosage scheme to the requirements of the younger population. Total number and time of administration of boluses and time to awakening were registered and used as criteria to adjust the dosage scheme. Predicted infusion rates are high (e.g., 24 mg/kg/hour for the first 10 minutes in neonates) and should be used cautiously. Delayed awakening, hypotension, and an increased incidence of bradycardia were reported in neonates and infants (81). Propofol can cause profound hypotension in neonates and pharmacokinetic–pharmacodynamic relationships in this age group remain elusive (82).
Pain on injection of propofol is a major problem occurring in up to 85% of children when no remedial action is taken (83). It can be minimized by injecting into a large vein, injecting the solution slowly, and administering at least 0.2 mg per kg of lidocaine immediately before, or with, the propofol (84). The use of a new propofol formulation in which the lipid component of long-chain triglycerides in the standard propofol formulation has been replaced by a mixture of long- and medium-chain triglycerides has been found to abolish pain in only 33% of children compared with 61% of children given the standard formulation mixed with lidocaine (85).
The vasodilator effects of propofol are greater than those of thiopental. Pediatric studies have consistently demonstrated a reduction in systolic and mean arterial pressures ranging from 5–30% occurring in the first 5 min following injection of propofol (58,60,86). Heart rate changes are variable in older children, but in one study, heart rate decreased significantly more in toddlers after propofol than after thiopental (58).
CLINICAL PEARL Pediatric studies have consistently demonstrated a reduction in systolic and mean arterial pressures (MAPs) ranging from 5% to 30% occurring in the first 5 minutes following injection of propofol (58,60,86).
The use of propofol for prolonged sedation in pediatric intensive care units (PICUs) is associated with a rare syndrome comprising metabolic acidosis, heart failure, lipemia, rhabdomyolysis, and death. The cause of this is unknown, but recent attention has focused on impairment of fatty acid oxidation by propofol as a possible cause (87,88). The fact that this propofol infusion syndrome is more common in children than in adults may be a reflection of the higher dose requirement for propofol in children. In view of these reports, the drug regulatory authorities of the United Kingdom, United States, and Canada have advised that propofol is contraindicated for intensive care sedation of children (89).
OPIOIDS
Opioid agonists produce analgesia and respiratory depression by combining with µ and κ opioid receptors. Until the late 1980s, the use of these drugs was avoided in neonates and young infants because of their increased susceptibility to respiratory depression. However, attitudes changed following the demonstration that neonates mount a massive stress response to surgery that can be modified by opioids (90). The susceptibility of young infants to opioid-induced respiratory depression may be due to increased penetration of the CNS by opioids, reduced capacity for opioid elimination, and developmental changes in the relative proportions and affinities of opioid receptors. However, respiratory depression measured by carbon dioxide response curves or by arterial oxygen tension are similar from 2 to 570 days of age at the same morphine blood concentration (91). Similar results are reported for fentanyl (92), suggesting that pharmacokinetic differences between term neonates and children play a major role. The introduction of newer opioid drugs with short context-sensitive half-times has rekindled interest in the use of opioids to avoid the major cardiovascular effects of volatile agents.
MORPHINE
Following an intravenous dose of morphine, brain uptake is slow due to poor lipid solubility. An equilibration half-time (T1/2keo) of 16 to 23 minutes is reported in adults (93,94). Similarly, the decay of CNS concentration is slow as it depends on elimination of the drug by the liver. Conjugation with glucuronide (UGT2B7) produces both active (morphine-6-glucuronide) and inactive metabolites (morphine-3-glucuronide), which are excreted by the kidneys.
Empiric studies have taught us that a concentration range (10 to 20 µg/L) has an analgesic effect (95) without associated adverse effects such as the respiratory depression observed with higher concentrations (91) or postoperative nausea and vomiting reported with higher doses (96). Fears of adverse effects, particularly respiratory depression, dictate that morphine dose is titrated to gain satisfactory analgesia (97,98). A regimen such as a loading dose of 50 µg per kg followed by 25 µg per kg at 5 minute intervals to control pain is a satisfactory method (99). A smaller dose of 20 µg per kg is used in neonates. The drug remains remarkably safe when used as an infusion in hospital practice. The overall incidence of serious harm was only 1:10, 000 with opioid infusion techniques, and those predisposed to harm can be identified (e.g., young infants, those with neurodevelopmental, respiratory, or cardiac comorbidities) (100).
Infusion regimens are based on clearance. Clearance increases from 3.2 L/hour/70 kg at 24 weeks PMA to 19 L/hour/70 kg at term, reaching adult values (80 L/hour/70 kg) at 6 to 12 months (see Fig. 2.6) (101,102). Oral bioavailability is approximately 35%. Infusion regimens that achieve a plasma concentration of 10 µg per L can be predicted from clearance (see Fig. 2.15). Morphine exhibits perfusion-limited clearance, and positive pressure ventilation, by reducing hepatic blood flow, also reduces clearance (102). While hepatic failure may be an obvious cause for reduced clearance, it is little appreciated that renal failure may also reduce clearance by approximately 30%. The kidney is also a major contributor to morphine glucuronide conjugation (103–106). Reduced morphine clearance has also been described in children with cancer (107,108), following cardiac surgery (109), and in critically ill neonates requiring extracorporeal membrane oxygenation (ECMO) (110,111), although contributions from hepatic failure, renal compromise, or positive pressure ventilation toward these reduced clearances were not acknowledged. Fears of respiratory depression dictate that infants aged less than 6 months receiving morphine infusions should be nursed in a high-dependency unit and monitored with continuous pulse oximetry (112,113).
FENTANYL
Fentanyl is a synthetic opioid, with lipid solubility about 600 times greater than that of morphine. High lipid solubility confers increased potency, rapid onset (T1/2keo 6.6 minutes in adults), and short duration of action. Fentanyl is a potent μ-receptor agonist with a potency 100 times greater than that of morphine. A plasma concentration of 15 to 30 µg per L is required to provide TIVA in adults, whereas the EC50, based on EEG evidence, is 10 μg per L (114,115). After a dose of 1 to 2 μg per kg, the clinical effects of fentanyl are terminated by redistribution, and its duration of action is limited to 20 to 30 minutes. However, after repeated doses or a continuous infusion, progressive saturation of peripheral compartments will result in prolonged duration of action.
Fentanyl is metabolized by oxidative N-dealkylation (CYP3A4) into norfentanyl and hydroxylated fentanyl. Clearance in preterm neonates is markedly reduced (T1/2β is 17.7 hours), contributing to prolonged respiratory depression in that population. The clearance of fentanyl is reduced to 70% to 80% of adult values in term neonates and, when standardized using allometry, reaches adult values (~50 L/hour/70 kg) within the first 2 weeks of life (7). Clearance of fentanyl in older infants (greater than 3 months of age) and children is greater than in adults when expressed as per kilogram (30.6 mL/kg/minute vs. 17.9 mL/kg/minute, respectively), resulting in a reduced elimination half-life (T1/2β 68 minutes vs. 121 minutes, respectively). These age-related changes follow the pattern portrayed in Figure 2.10. Fentanyl clearance may be impaired with decreased hepatic blood flow (e.g., from increased intra-abdominal pressure in neonatal omphalocele repair); a maldistribution of blood away from regions of concentrated cytochrome enzyme activity in the liver may also play a role (116). Fentanyl’s volume of distribution at steady state (Vdss) is approximately 5.9 L per kg in term neonates and decreases with age to 4.5 L per kg during infancy, 3.1 L per kg during childhood, and 1.6 L per kg in adults (117). Chest wall and glottic rigidity have been reported after IV administration of all opioids, although most commonly after fentanyl.
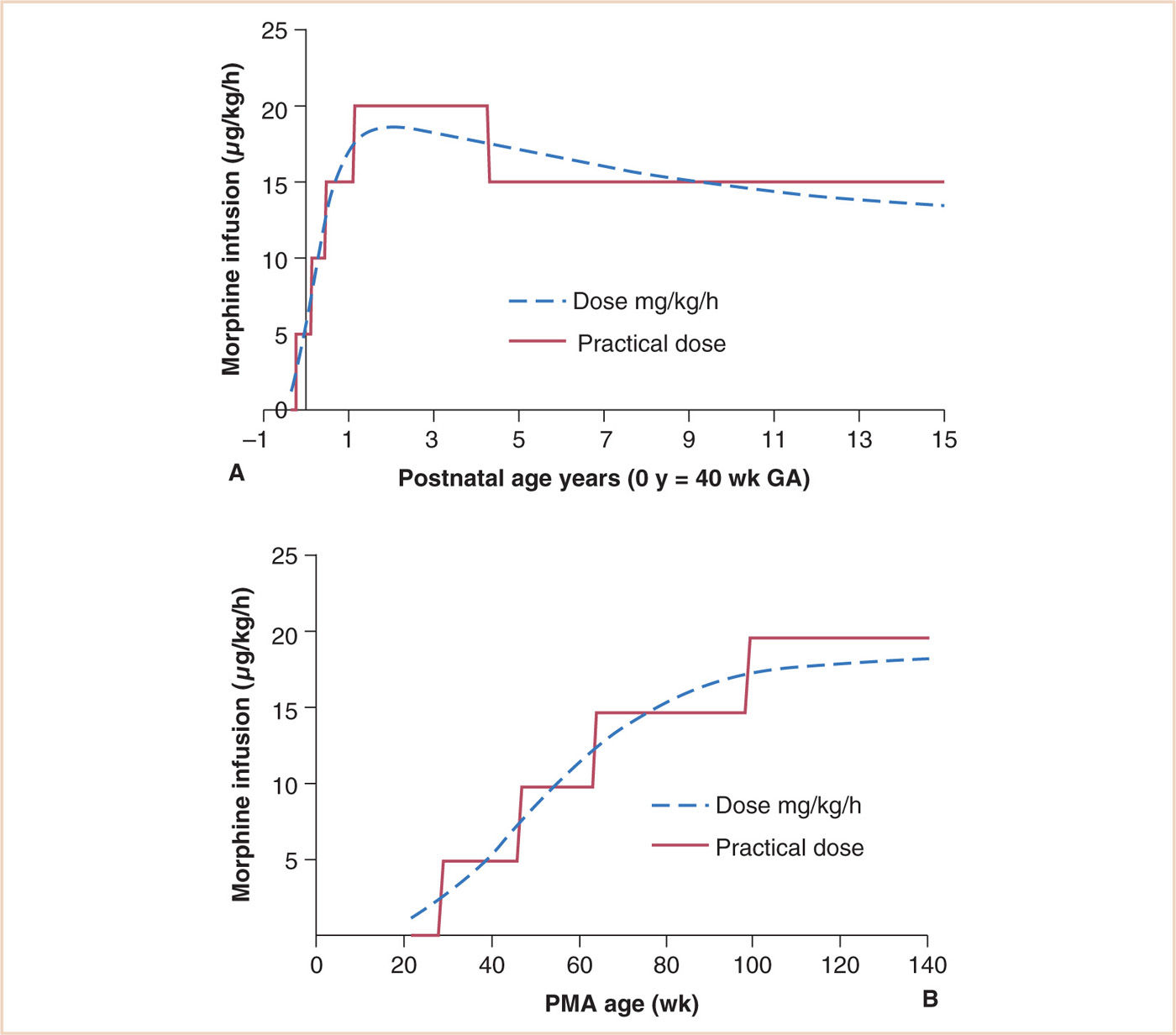
FIGURE 2.15 Age-based infusion dosing for morphine with a target average steady state concentration of 10 μg per L in children. The dotted line is the prediction based on age and typical weight for age. The solid line is the suggested practical infusion rate dose in μg/kg/hour at different postnatal ages (PNAs). A: Dose related to PNA. B: Dose related to PMA.
(From Anderson BJ, Holford NH. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child. 2013;98:737–744, with permission.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


