Developmental Neurobiology, Neurophysiology, and the Picu
Larry W. Jenkins
Patrick M. Kochanek
KEY POINTS
 Important biochemical, molecular, physiologic, and structural differences in central nervous system (CNS) development influence treatment of brain injury in newborns, infants, children, adolescents, and adults.
Important biochemical, molecular, physiologic, and structural differences in central nervous system (CNS) development influence treatment of brain injury in newborns, infants, children, adolescents, and adults. A unique aspect of programmed cell death (PCD) in the developing brain versus the adult brain is an imbalance between pro-death and pro-survival pathways induced by decreases in trophic and neurotransmitter neuronal input. Thus, the developing brain may be more sensitive to injuries that mimic regional developmental changes in trophin and neurotransmitter levels. It may also be sensitive to anesthetics and/or sedatives as suggested by recent studies in primates. This is a vital area for current and future research to define the potential risks for infants in the PICU.
A unique aspect of programmed cell death (PCD) in the developing brain versus the adult brain is an imbalance between pro-death and pro-survival pathways induced by decreases in trophic and neurotransmitter neuronal input. Thus, the developing brain may be more sensitive to injuries that mimic regional developmental changes in trophin and neurotransmitter levels. It may also be sensitive to anesthetics and/or sedatives as suggested by recent studies in primates. This is a vital area for current and future research to define the potential risks for infants in the PICU. Synaptogenesis is an active process that has a high rate of occurrence until puberty.
Synaptogenesis is an active process that has a high rate of occurrence until puberty. Myelination persists longer than most other CNS developmental processes, continuing through adolescence.
Myelination persists longer than most other CNS developmental processes, continuing through adolescence. Around the time of birth, the neurotransmitter g-aminobutyric acid (GABA) switches from being excitatory to inhibitory in the CNS. The developing brain prior to this switch has enhanced sensitivity to excitotoxic injury.
Around the time of birth, the neurotransmitter g-aminobutyric acid (GABA) switches from being excitatory to inhibitory in the CNS. The developing brain prior to this switch has enhanced sensitivity to excitotoxic injury. The lower limits of blood pressure autoregulation of cerebral blood flow (CBF) for infants and adults are similar, putting the developing brain at risk for ischemia, as normal mean arterial pressure is age dependent.
The lower limits of blood pressure autoregulation of cerebral blood flow (CBF) for infants and adults are similar, putting the developing brain at risk for ischemia, as normal mean arterial pressure is age dependent. CBF and cerebral metabolic rate increase from birth and plateau between 3 and 9 years of age. This developmental progression mirrors synapse formation in the brain.
CBF and cerebral metabolic rate increase from birth and plateau between 3 and 9 years of age. This developmental progression mirrors synapse formation in the brain. Cerebrovascular tight junctions prevent movement of hydrophilic substances, osmotic agents, and proteins across the blood-brain barrier (BBB).
Cerebrovascular tight junctions prevent movement of hydrophilic substances, osmotic agents, and proteins across the blood-brain barrier (BBB).The field of pediatric neurointensive care is challenged with designing and implementing optimal therapies for complex insults, such as traumatic brain injury (TBI), asphyxial arrest, stroke, refractory status epilepticus, and infections, for the most complex organ system—the central nervous system (CNS). The challenge is magnified in pediatrics by the need to accomplish this goal in an optimal manner, whether the patient is a newborn, infant, child, or adolescent. Salient differences in structural, biochemical, physiologic, and behavioral components of the brain from infancy to adolescence have been recognized, and an understanding of these factors may be important to the clinician when appropriately defining  prognosis and/or guiding therapy. This chapter provides fundamental insights into this complexity and is organized around key concepts and developmental milestones that should provide useful constructs for understanding both developmental brain function and pathology in clinical practice. It provides basics in developmental neurobiology and neurophysiology that will help the reader navigate the chapters that follow. We recognize that, in several areas, a complete picture of developmental differences in neurobiology and neurophysiology across the PICU-relevant age spectrum is lacking—and extrapolation from data on newborns or adults is necessary. In some cases, only data from experimental animal models are available. Nevertheless, certain stages of development may be particularly resistant or vulnerable to PICU-relevant insults that may interfere with subsequent normal developmental progression. Key factors germane to pediatric intensive care that differentiate the newborn, infant, child, adolescent, and adult brain are discussed whenever possible. For more details, the reader is referred to several excellent reviews (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20) to which much of this chapter is indebted.
prognosis and/or guiding therapy. This chapter provides fundamental insights into this complexity and is organized around key concepts and developmental milestones that should provide useful constructs for understanding both developmental brain function and pathology in clinical practice. It provides basics in developmental neurobiology and neurophysiology that will help the reader navigate the chapters that follow. We recognize that, in several areas, a complete picture of developmental differences in neurobiology and neurophysiology across the PICU-relevant age spectrum is lacking—and extrapolation from data on newborns or adults is necessary. In some cases, only data from experimental animal models are available. Nevertheless, certain stages of development may be particularly resistant or vulnerable to PICU-relevant insults that may interfere with subsequent normal developmental progression. Key factors germane to pediatric intensive care that differentiate the newborn, infant, child, adolescent, and adult brain are discussed whenever possible. For more details, the reader is referred to several excellent reviews (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20) to which much of this chapter is indebted.
prognosis and/or guiding therapy. This chapter provides fundamental insights into this complexity and is organized around key concepts and developmental milestones that should provide useful constructs for understanding both developmental brain function and pathology in clinical practice. It provides basics in developmental neurobiology and neurophysiology that will help the reader navigate the chapters that follow. We recognize that, in several areas, a complete picture of developmental differences in neurobiology and neurophysiology across the PICU-relevant age spectrum is lacking—and extrapolation from data on newborns or adults is necessary. In some cases, only data from experimental animal models are available. Nevertheless, certain stages of development may be particularly resistant or vulnerable to PICU-relevant insults that may interfere with subsequent normal developmental progression. Key factors germane to pediatric intensive care that differentiate the newborn, infant, child, adolescent, and adult brain are discussed whenever possible. For more details, the reader is referred to several excellent reviews (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20) to which much of this chapter is indebted.CENTRAL NERVOUS SYSTEM DEVELOPMENT
Brain Development Timeline
CNS development occurs through the process of neurulation during embryogenesis. The neural plate is formed by ectodermal tissue at ˜2 weeks of gestation. By the 18th day of gestation, the neural plate forms the neural groove, which in turn eventually fuses by the 3rd gestational week, giving rise to the neural tube. Completed human neural tube formation occurs between 26 and 28 gestational days (18). Throughout the first month of human gestation, specific CNS regions, such as the forebrain, midbrain, and hindbrain, form because of neurogenesis and cellular migration. Concomitant with regional CNS development, neurogenesis and proliferation, migration, differentiation, synaptogenesis, apoptosis, and myelination occur and continue postnatally up to 10 years of age (Fig. 55.1). Major CNS developmental milestones in gestational weeks are as follows (12):
3-4 weeks—Formation of the neural tube occurs.
5-10 weeks—Hemispheres form.
8-18 weeks—Neuronal proliferation is ongoing.
12-24 weeks-Neuronal migration proceeds.
25+ weeks-Neuronal arborization, synaptogenesis, programmed neuronal death, and neural connectivity occur.
40+ weeks-Myelination is ongoing.
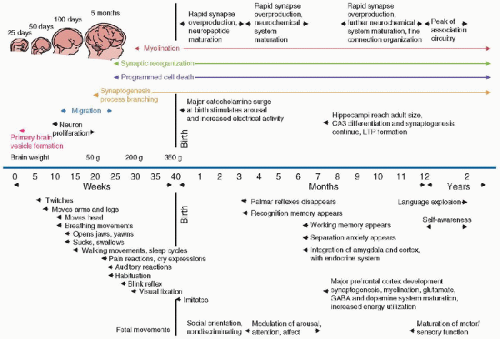 FIGURE 55.1. The key events of the human developmental timeline. The appearance of specific structural and functional developmental events is shown for the human fetus, infant, and young child up to 2 years of age. LTP, long-term potentiation; GABA, g-aminobutyric acid. (Modified from Lagercrantz H, Ringstedt T. Organization of the neuronal circuits in the central nervous system during development. Acta Paediatr 2001;90:707-15; additional data from Levitt P. Structural and functional maturation of the developing primate brain. J Pediatr 2003;143:S35-45; and from Herschkowitz N. Neurological bases of behavioral development in infancy. Brain Dev 2000;22:411-16.) |
Development of Specific Brain Regions
Human brain size increases dramatically, beginning from early gestation and continuing for at least 2-3 postnatal years. CNS growth, based on changes in gross brain size, peaks at around 4 months postnatally. However, specific brain regions have different growth time windows (Fig. 55.2) and periods of genetic and environmental vulnerabilities before and after birth (18). In general, the forebrain develops slower than the hindbrain, with the medial aspects of the hindbrain developing faster than the lateral. The neocortical and hippocampal structures grow mostly during the fetal period but do have some continuing neuronal and glial postnatal development. In contrast, the thalamus and hypothalamus develop during the early fetal and late embryonic periods, as does the mesencephalon. The pons and medulla of the hindbrain develop primarily during the embryonic period, which in humans encompasses weeks 3-7.5 of gestation (21).
Neurogenesis and Proliferation
The neural tube is formed by the neuroectoderm, with neuroepithelial cells differentiating into various types of neurons and glia. The mature human brain contains an estimated 1010 neurons (22). Neural precursor cells from the epithelium form the developing CNS and undergo mitotic arrest at various times during development. Prior to becoming postmitotic cells, these cells determine their position within the embryonic axis and either reenter the cell cycle to increase the precursor pool or enter mitotic arrest (7). During development, neuronal populations within each brain region are highly regulated along a structured timetable. This regional variability appears conserved across species with regard to neuronal developmental patterns, but with species-specific timelines. For example, neurogenesis occurs within days in the rat compared to weeks in human. Environmental or genetic stress during neurogenesis is a particularly vulnerable time for regional brain development. However, the CNS is more resistant to such stressors following neurogenesis. Fetal alcohol syndrome is a classic example of environmental stress that can interfere with the developmental process of neurogenesis (18).
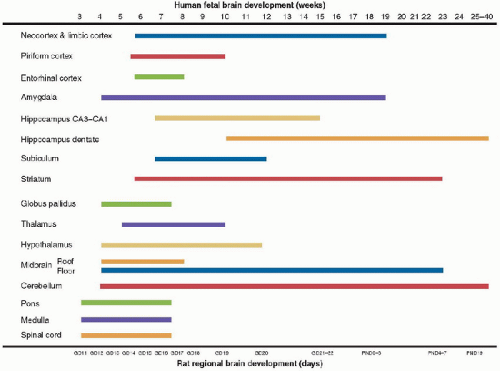 FIGURE 55.2. Human fetal brain development. The developmental timeline of selected human brain regions is shown in comparison with rat development timeline. The rat is the most commonly used species to study both normal and injury-related CNS development and thus has the most extensive normative database. GD, gestational day; PND, postnatal day. (Modified from Rice D, Barone S, Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ Health Perspective 2000;108(suppl 3):551-33, with permission.) |
The prenatal stage of brain development is characterized by rapid cell division under the control of the cell cycle and, in turn, numerous cell signaling pathways, including at least nine different growth factor cascades (4). Cells also must duplicate their organelles and cellular molecules to maintain their size; thus, growth processes must also be coordinated with cellular replication. It has been estimated that ˜200,000 new neurons are produced each minute at between 8 and 18 weeks of gestation. In contrast, it has been proposed that little neurogenesis occurs after birth, except in some select brain regions that continue into adulthood (12).
Programmed Cell Death
Neuronal cell type and number are regulated by apoptotic cell death during CNS development, which occurs in waves. Programmed cell death (PCD) begins in zones of proliferation and recurs as CNS remodeling proceeds based on the kind and number of connections made by each individual neuroblast and neuron (4). Furthermore, PCD persists postnatally owing to continued CNS development. Two types of developmental PCD have been classified: (a) a proliferative apoptosis that affects morphogenetic processes involving neural precursor cells and postmitotic neuroblasts, and (b) a neurotrophicrelated apoptosis that affects postmitotic neurons that fail to establish appropriate synaptic connections. Apoptotic regulation of neurons that undergo PCD prior to developing synaptic contacts (proliferative apoptosis) has been proposed to differ from target-dependent neuronal death pathways (neurotrophic-related apoptosis) (7) (Fig. 55.3). Proliferative neuronal apoptosis prevents premature and dysfunctional neurogenesis from occurring secondary to premature differentiation signals.
Interference in normal developmental neuronal death cascades by environmental or genetic stress can result in a number of different pathologies (4). In contrast to the trophinmediated signals that promote growth and survival, extracellular signaling proteins inhibit these processes. These survival and death signals, as will be seen in Chapter 56, appear to play a key role in the evolution of neuronal death after CNS injuries such as asphyxia, TBI, seizures, infections, and other  PICU-relevant insults.
PICU-relevant insults.
PICU-relevant insults.The Bcl-2 protein family, the adaptor protein Apaf1, and the cysteine protease caspase family are the principal regulators of PCD (7). PCD pathways involve both intrinsic and extrinsic
types of neuronal death. Intrinsic PCD signaling pathways involve a reduction in mitochondrial membrane potential, resulting in the release of cytochrome C by the activation of Apaf1 and caspase 9, with ultimate activation of caspase 3. Bcl-2 family proteins Bcl-2 and Bcl-XL are pro-survival molecules that inhibit the release of mitochondrial cytochrome C, whereas pro-death molecules, such as Bax and Bad, can directly affect mitochondrial membrane integrity.
types of neuronal death. Intrinsic PCD signaling pathways involve a reduction in mitochondrial membrane potential, resulting in the release of cytochrome C by the activation of Apaf1 and caspase 9, with ultimate activation of caspase 3. Bcl-2 family proteins Bcl-2 and Bcl-XL are pro-survival molecules that inhibit the release of mitochondrial cytochrome C, whereas pro-death molecules, such as Bax and Bad, can directly affect mitochondrial membrane integrity.
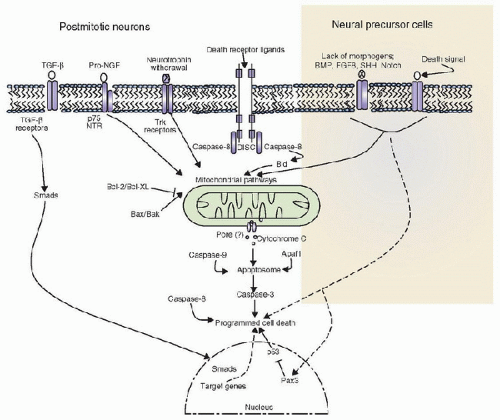 FIGURE 55.3. Both neural precursor cells and postmitotic neurons utilize mitochondrial pathways (cytochrome C and apoptosome) in programmed cell death (PCD). Bcl-2 family proteins (Bcl-2, Bcl-X, Bax, and Bak) modulate and regulate the mitochondrial PCD pathway. Receptormediated PCD via caspase 8 has also been shown to participate in postmitotic neurons but not in neural precursor cells, which undergo PCD via the withdrawal of morphogens and regulation of the p53 pathway by the Pax3 transcription factor. Pax3 is a transcription factor that regulates neural development at the transcriptional level and can modulate the cell cycle via p53 to modulate PCD pathways. TGF-β and proNGF also stimulate PCD in postmitotic neurons. Smad proteins are regulators of transcription that are phosphorylated by activated TGF-β receptors and, in turn, activate gene expression patterns that modulate PCD and survival. (Modified from De Zio D, Giunta L, Corvaro M, et al. Expanding roles of programmed cell death in mammalian neurodevelopment. Semin Cell Dev Biol 2005;16:281-94, with permission.) |
In the extrinsic pathway, removal of neurotrophic factors leads to activation of the protein kinase c-Jun kinase (JNK). JNK can phosphorylate different substrates, including c-Jun, which, in turn, activates transcription of Fas-L by binding to AP-1 sites in the gene promoter site (23). Fas modulates various effectors to activate caspases directly, to activate the stressactivated JNK pathway, or to inhibit the protein kinase B (PKB or Akt) survival pathway (23). Other receptor-coupled prodeath pathways also exist in developmental CNS PCD, but the mitochondrial apoptotic pathway is the most important (7) (Fig. 55.3). As many as 70% of developing neurons die via PCD during embryogenesis to eliminate excess cell numbers and to assist in neural tube closure. Due to the upregulation of PCD machinery during development, the developing CNS may be more prone to injury-related PCD than is the adult brain. An especially sensitive age for injury-induced CNS PCD is in newborn infants in the PICU environment, discussed in greater detail in Chapter 56.
Migration, Differentiation, and Axonal Guidance
Neuronal migration occurs at between 12 and 24 gestational weeks in humans and is modulated by neurotransmitters such as glutamate. Importantly, glutamate N-methyl-D-aspartate (NMDA) receptor antagonists can inhibit neuronal migration and affect developmental neuronal apoptosis (12), which may have important effects on developing CNS recovery and plasticity and, therefore, great relevance to the PICU.
The developmental processes responsible for the progression of neural lineages from stem cells to progenitors to postmitotic precursors and, ultimately, to mature neurons are controlled by multiple pathways (24). Stem cell commitment to neuronal lineage and neuronal progenitor specification to a specific neural subtype are two distinct steps of neurogenesis. However, they are linked mechanistically by the same genes that regulate both processes (24). Both extracellular and intracellular signals modulate new gene expression that, in large part, determines neuronal and glial phenotypes. Differentiated neurons possess unique and characteristic sizes, shapes, polarities, and expressions of neurotransmitters, neurotrophins, and receptors, to name but a few differentiation attributes. Knowledge of this area of research is important, as both positive and negative experimental results have been reported concerning
the potential of stem cells as therapy after brain injury, and this is sure to remain an active field of inquiry in the future.
the potential of stem cells as therapy after brain injury, and this is sure to remain an active field of inquiry in the future.
Four primary types of signals guide axonal growth and target contact: chemoattractant, chemorepellent, contact attractive, and contact repellent. The first two classes of molecules are diffusible molecules that act over longer distances, while the latter two are within the extracellular matrix or membrane. Axonal pathfinding is most dependent on repulsive cues and pioneer axons that reach targets early in development. Some of these guidance molecules also affect cell migration, as both developmental processes tend to share common mechanisms (12).
Synaptogenesis, Gliogenesis, and Myelination
Synaptogenesis is one of the most important developmental processes that occur during childhood and is an important potential mechanism of CNS injury and recovery in PICU-relevant injuries. Based on nonhuman primate studies, a tentative timetable composed of five temporally distinct phases of synaptogenesis has been proposed for human development (12)  (Figs. 55.1 and 55.4). At ˜6-8 weeks of gestation, phase 1 of synaptogenesis is limited to the subplate. Beginning at ˜12-17 weeks of gestation, synaptogenesis phase 2 remains somewhat limited to the cortical plate, with most new synapses on neuronal dendritic shafts. Phase 3 is more rapid and dynamic; with up to 40,000 new synapses made per second, it begins at ˜20-24 weeks of gestation and lasts up to 8 months postnatally. Similarly, phase 4 has a high rate of synaptogenesis, lasting until puberty. The third and fourth phases are more influenced by use-dependent experience. Lastly, phase 5 levels of synaptogenesis persist throughout adulthood to age 70, but with significant synapse loss during this period as well (12).
(Figs. 55.1 and 55.4). At ˜6-8 weeks of gestation, phase 1 of synaptogenesis is limited to the subplate. Beginning at ˜12-17 weeks of gestation, synaptogenesis phase 2 remains somewhat limited to the cortical plate, with most new synapses on neuronal dendritic shafts. Phase 3 is more rapid and dynamic; with up to 40,000 new synapses made per second, it begins at ˜20-24 weeks of gestation and lasts up to 8 months postnatally. Similarly, phase 4 has a high rate of synaptogenesis, lasting until puberty. The third and fourth phases are more influenced by use-dependent experience. Lastly, phase 5 levels of synaptogenesis persist throughout adulthood to age 70, but with significant synapse loss during this period as well (12).
(Figs. 55.1 and 55.4). At ˜6-8 weeks of gestation, phase 1 of synaptogenesis is limited to the subplate. Beginning at ˜12-17 weeks of gestation, synaptogenesis phase 2 remains somewhat limited to the cortical plate, with most new synapses on neuronal dendritic shafts. Phase 3 is more rapid and dynamic; with up to 40,000 new synapses made per second, it begins at ˜20-24 weeks of gestation and lasts up to 8 months postnatally. Similarly, phase 4 has a high rate of synaptogenesis, lasting until puberty. The third and fourth phases are more influenced by use-dependent experience. Lastly, phase 5 levels of synaptogenesis persist throughout adulthood to age 70, but with significant synapse loss during this period as well (12).As a developing neuron reaches its destined position in the CNS, it extends dendrites and axons to distant targets via growth cones, which are guided by numerous extracellular molecules. On making target contact, presynaptic neurotransmitters or other secreted molecules diffuse and bind to the postsynaptic membrane and, along with other signals, induce postsynaptic receptor clustering and other elements of the synapse. Consequently, numerous proteins, molecules, and signal cascades play a role in synaptogenesis (22). Synaptic vesicle accumulation in the presynaptic terminal of a brain region is a helpful hallmark of the level of synapse maturation (25). A tremendous amount of synaptogenesis is ongoing in young children that could have important implications for injury response and recovery in the PICU.
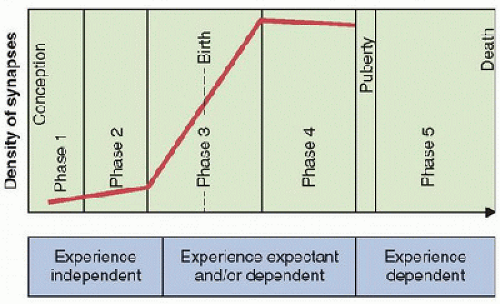 FIGURE 55.4. Synaptic changes (shown as relative densities based on a logarithmic scale) from data of the macaque monkey that has been extrapolated to humans. Phases of synaptogenesis that are predominately experience dependent (require neural activity and are epigenetically regulated) or independent (genetically programmed) are indicated by the boxes along the X-axis. (Modified from Lagercrantz H, Ringstedt T. Organization of the neuronal circuits in the central nervous system during development. Acta Paediatr 2001;90:707-15, with permission.) |
Glia are not passive participants in CNS development; rather, they exert significant influence over neuronal development. Glia include astrocytes, oligodendrocytes, radial glia, and microglia. Astrocytic development occurs well after neuronal migration and differentiation, and oligodendrocytic development occurs after that of axonogenesis (18). Astrocytes have a multitude of functions in the CNS aside from the structural support of neurons. These functions include K+ buffering, H+ and Ca2+ ion homeostasis, ammonia detoxification, free radical scavenging, metal sequestration, growth factor production, immune response participation, and neuronal metabolic support functions (pH regulation, neurotransmitter uptake, supply of glycogen and tricarboxylic acid cycle intermediates, and provision of neurotransmitter precursors). Astrocytes also participate in synaptogenesis and neurogenesis in CNS development as well as in cognitive function (17). Protoplasmic astrocytes occur in gray matter, and fibrous astrocytes in white matter. An important feature of astrocytes is the use of the energy-dependent Na+ gradient to uptake glutamate and K+ and to regulate H+ ions. As a result, excessive release of glutamate, energy failure, neuronal depolarization, or tissue acidosis in brain injury result in astrocytic swelling in both the perivascular and perineuronal astrocytic compartments, which can increase diffusion distances in the brain for metabolic gases (O2 and CO2), substrates, and waste products. It has been proposed that severe glial swelling may compress capillaries to reduce cerebral blood flow (CBF) and distort synaptic contacts, resulting in neuronal synaptic deafferentation. These changes can become important in a variety of CNS insults (see Fig. 56.12).
Microglia are CNS immune cells of myeloid origin that are similar to macrophages and represent 10% of the CNS cell populations in adults (19). They are normally in a resting state in normal brain, but with activation in response to infection or CNS injury, they morphologically (enlarge) and functionally change, resulting in the upregulation of cytokines and chemokines, as well as surface antigens. They have been implicated in the response to encephalitis, ischemia, TBI, and demyelinating diseases.
Oligodendrocytes produce myelin, modulate axonal function in the CNS, and are vulnerable to excitotoxic injury, decreased trophin levels, and oxidative stress conditions. Neurons and oligodendrocytes signal each other during myelination to modulate neurofilament spacing, phosphorylation, and axonal diameter. As myelination continues to occur until 10 years of age, injury to oligodendrocytes and altered myelination is an important consideration in the PICU in children who suffer brain injuries.
Myelination persists longer than most other CNS developmental processes, continuing through adolescence, making it, like synaptogenesis, another aspect of CNS development of  special relevance to the PICU infant or child (18) (Fig. 55.1). In fact, myelination continues in humans until at least 30 years of age, and new evidence suggests that myelination may even be an important mechanism of activity-dependent plasticity (defined as both short- and long-term changes in synaptic strength stimulated by altered neural electrical activity via altered synaptic protein levels, protein posttranslational modifications, and nerve conduction velocity) (26). Several lines of evidence suggest that myelination is a process that is influenced by environmental factors. Neural activity also affects myelination. White matter development in children correlates with motor skill and increased cognitive function. MRI has shown a strong correlation between white matter development and cognitive function (intelligence quotient) in children and
special relevance to the PICU infant or child (18) (Fig. 55.1). In fact, myelination continues in humans until at least 30 years of age, and new evidence suggests that myelination may even be an important mechanism of activity-dependent plasticity (defined as both short- and long-term changes in synaptic strength stimulated by altered neural electrical activity via altered synaptic protein levels, protein posttranslational modifications, and nerve conduction velocity) (26). Several lines of evidence suggest that myelination is a process that is influenced by environmental factors. Neural activity also affects myelination. White matter development in children correlates with motor skill and increased cognitive function. MRI has shown a strong correlation between white matter development and cognitive function (intelligence quotient) in children and
adolescents (26). While such data are correlative and do not prove an underlying relationship, it has been postulated that increased white matter conduction speed may have as strong an influence on synaptic amplitude as do changes in pre- and post-synaptic elements and neurotransmitter function in plasticity paradigms, such as long-term potentiation (LTP), an electrophysiologic model of memory formation (26). Furthermore, neural activity may produce activity-dependent interactions between myelinating glia, neurons, and myelinated axons (26).
special relevance to the PICU infant or child (18) (Fig. 55.1). In fact, myelination continues in humans until at least 30 years of age, and new evidence suggests that myelination may even be an important mechanism of activity-dependent plasticity (defined as both short- and long-term changes in synaptic strength stimulated by altered neural electrical activity via altered synaptic protein levels, protein posttranslational modifications, and nerve conduction velocity) (26). Several lines of evidence suggest that myelination is a process that is influenced by environmental factors. Neural activity also affects myelination. White matter development in children correlates with motor skill and increased cognitive function. MRI has shown a strong correlation between white matter development and cognitive function (intelligence quotient) in children and adolescents (26). While such data are correlative and do not prove an underlying relationship, it has been postulated that increased white matter conduction speed may have as strong an influence on synaptic amplitude as do changes in pre- and post-synaptic elements and neurotransmitter function in plasticity paradigms, such as long-term potentiation (LTP), an electrophysiologic model of memory formation (26). Furthermore, neural activity may produce activity-dependent interactions between myelinating glia, neurons, and myelinated axons (26).
NEUROTRANSMITTER AND NEUROTROPHIN DEVELOPMENT
The synapse serves as the anatomical substrate for information flow between neurons and the point at which the release and response to neurotransmitters predominately occur within the nervous system (Fig. 55.5). The release of neurotransmitters from within presynaptic vesicles occurs by fusion with the presynaptic plasma membrane due to electrical depolarization and the influx of Ca2+ into the presynaptic bouton, which contains synaptic vesicles. Receptors in the postsynaptic membrane couple directly to ion channels or second messengers to mediate downstream effects. The removal of neurotransmitters that have been released occurs by glial uptake, enzymatic degradation, or transport proteins coupled to the Na+ gradient established by synaptic and glial sodium and potassium adenosine triphosphate translocase enzymes (Na/K-ATPase). Neurotransmitter levels and receptor expression serve critical roles in synapse formation and in the circuitry and networks necessary for behavioral function in the immature and mature CNS. Furthermore, synaptic activity mediated by neurotransmitters is a requirement for the survival of developing synaptic contacts in the immature brain (9). It is well documented that neurotransmitters both mediate synaptic transmission and have trophic functions (5,9,18). Neurotransmitters and neuromodulators that have been shown to play important roles in development include glutamate, γ-aminobutyric acid (GABA), acetylcholine, catecholamines, serotonin, and opioids. The human developmental neurotransmitter timeline displays considerable regional and temporal variation for various transmitter systems (18) (Figs. 55.6 and 55.7). These variations come into play when considering age- and regional-dependent injury, manipulations of the transmitter systems, and the possible effects of the transmitter systems on recovery. Insults to the brain at vulnerable developmental periods can produce longterm structural and functional CNS changes. Given the important neurotransmitter functions during CNS development, it is not surprising that a number of recent studies have implicated age-dependent adverse effects with the manipulation of neurotransmitter systems, especially the glutamatergic and GABAergic systems, using either commonly employed sedative and anesthetic agents or treatments with such agents after ischemia, status epilepticus, or TBI (5,27,28).
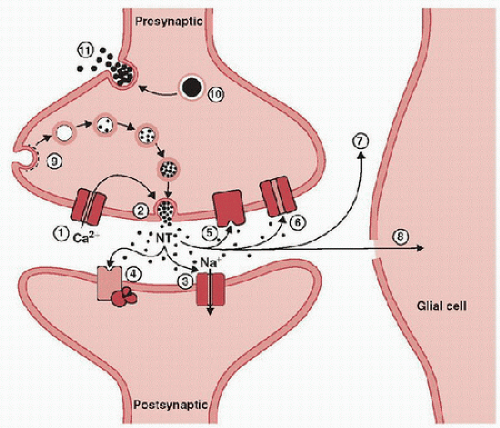 FIGURE 55.5. Schematic of a typical axodendritic synapse as seen at a dendritic spine. Calcium entry triggered by presynaptic terminal depolarization (1) induces neurotransmitter (NT)-containing synaptic vesicle exocytosis (2), which in turn, releases neurotransmitters into the synaptic cleft. NTs bind to their receptors at the postsynaptic density (concentration of postsynaptic proteins involved in synaptic function) of the postsynaptic membrane. These receptors modulate either ion channels (3) or G-proteins that stimulate second-messenger production (4). Similar NT presynaptic receptors (5) are also activated, which can modulate subsequent NT presynaptic release. The uptake of many NTs occurs via a transport protein coupled to the sodium gradient at the presynaptic terminal (e.g., dopamine, norepinephrine, glutamate, and GABA) (6) or by degradation (acetylcholine) (7) or uptake by glia (glutamate) (8). Synaptic vesicle membranes are recycled by clathrin-mediated endocytosis (9). Large, dense core vesicles (10) that store protein and neuropeptides are released by repetitive stimulation at a more distant site from the postsynaptic density (11). (From Holz RW, Fisher SK. Basic Neurochemistry. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999, with permission.) |
Preterm fetuses and newborn infants appear to be at particularly critical periods of developmental neurotransmitter vulnerability. Before birth, the majority of neuromodulators and neurotransmitters increase during synaptogenesis. At birth, increased brain activity and a surge of catecholaminergic activity are associated with arousal. A decrease in adenosine occurs, along with a desensitization of adenosine receptors during the
first postnatal days (9). In addition, periods of developmental “switches” in neurotransmitter function occur at birth, and sensitivity to glutamate toxicity may be especially high and vulnerable at this time.
first postnatal days (9). In addition, periods of developmental “switches” in neurotransmitter function occur at birth, and sensitivity to glutamate toxicity may be especially high and vulnerable at this time.
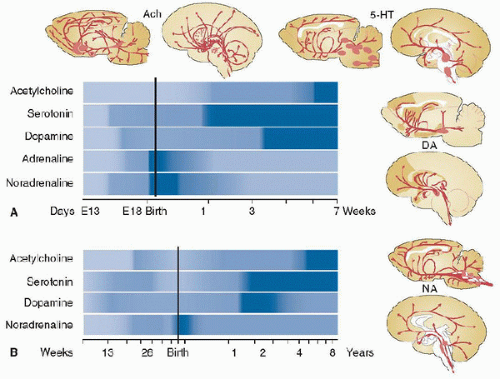 FIGURE 55.6. Comparison of rat (A) and human brain (B) timelines of the regional and temporal distribution of several major neurotransmitter levels during development. Ach, acetylcholine; 5-HT, serotonin; DA, dopamine; NA, noradrenaline. (From Herlenius E, Lagercrantz H. Development of neurotransmitter systems during critical periods. Exp Neurol 2004;190(suppl 1):S8-21. with permission.) |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


