FIGURE 18.1 Morphologic types of craniosynostosis. Diagrammatic views of different morphologic types of craniosynostosis are shown in the center of the figure. The premature closed suture is marked as a bold line. The sutures involved determine the shape of the skull. The diagrammatic view of each synostosis type is matched with the corresponding radiologic findings. The fused suture is seen as a hypertrophic keel or ridge on the x-rays.
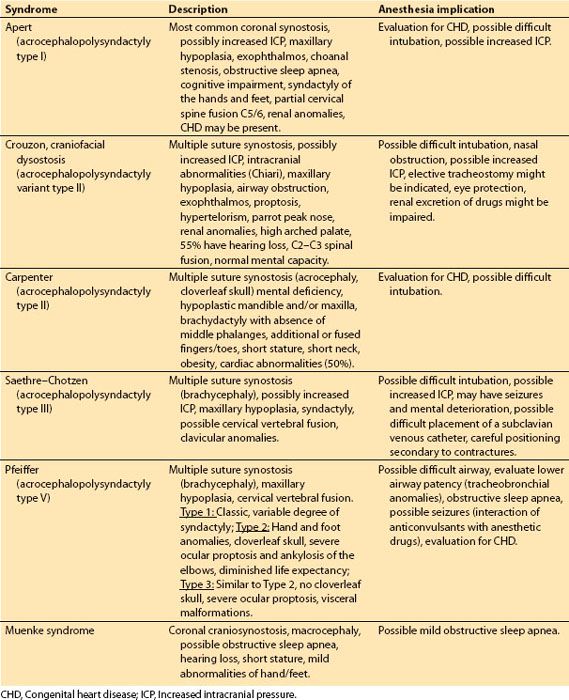
Table 18.1 Common craniofacial syndromes associated with craniosynostosis
b. Progressive postnatal craniosynostosis is characterized by a normal skull shape and normal radiologic study results at birth followed by midface hypoplasia and occasionally hypertelorism as the infant grows. Frequently, an association with the classic Crouzon syndrome phenotype occurs. These cases are significant because, although they have open sutures in infancy and do not initially display the physical manifestation of craniosynostosis, they develop multiple-suture craniosynostosis over time with slow development of symptoms of increased intracranial pressure (headaches, vomiting, irritability, papilledema, progressive optic atrophy, seizures, and/or bulging fontanels) that ultimately requires surgical interventions.
c. Shunting craniosynostosis. Shunting procedures in early infancy may result in overlapping of the calvarial bones and can induce secondary craniosynostosis.
d. Positional plagiocephaly also known as deformational plagiocephaly is a condition most commonly found in infants and is characterized by a flat spot on the back or one side of the head caused by remaining in one position for too long. These changes are not related to premature closure of cranial sutures and treated nonsurgically by orthotic treatment using a helmet if done early enough.
CLINICAL PEARL
In keeping with the general philosophy of the correction of congenital defects, there is consensus that surgical correction of craniosynostosis during early infancy is advantageous.
2. Surgical management. Craniofacial surgical treatments vary with age, location, and number of sutures involved. Many surgeons prefer to operate early in life to capitalize on the ameliorating effects of brain growth on skull shape. The following are some of the surgical management techniques:
a. Endoscopic strip craniectomy (ESC) is considered for infants (1 to 6 months) for all single suture and simple multiple suture craniosynostoses. ESCs are short surgical procedures (approximately 45 minutes) associated with small amount of blood loss, low incidence of blood transfusions, and ICU admissions. The average length of hospital stay is between 1 and 2 days with the majority of patients discharged on the first postoperative day [1].
Small skin incisions are performed perpendicular to the stenosed sutures. Cranial burr holes are made through the midline of each scalp incision. An endoscope is used to visualize the fused suture, identify emissary veins, dural attachments, and assure hemostasis. The closed suture is resected and removed through the burr hole.
CLINICAL PEARL
Less-invasive surgical techniques (e.g., ESC) are associated with a significant reduction in blood transfusions, intensive care unit (ICU) admissions, and costs. It remains to be determined whether the indications for less-invasive surgical techniques and open reconstruction procedures for single- and multiple-suture craniosynostosis can be better defined to further improve risk/benefit profile and costs.
b. Spring-assisted cranioplasty utilizes implanted cranial springs that allow gradual correction of the skull malformation over time and is used in infants older than 3 months of age. Optimal results are achieved in infants younger than 6 months of age while the skull still being relatively pliable. This technique involves an osteotomy and resection of the prematurely closed suture combined with the insertion of one to three stainless steel springs across the newly created false suture to allow ongoing dynamic correction of the skull anomaly over the following months. The ends of the stainless steel omega-shaped spring expanders are placed under tension into burr holes approximately 1 to 2 cm apart with the body of the spring bent to conform to the curvature of the skull. The expansion of a spring is a slow process and is monitored both clinically and radiologically. The springs are surgically removed after adequate cranial expansion [2].
c. Open reconstruction procedures represent a variety of surgical procedures that are usually performed in infants older than 6 to 12 months of age to limit operative morbidity and mortality. However, the deformities are more severe at this late stage. Open reconstruction procedures include simple strip craniectomy, p cranioplasties (paramedian strip craniectomies and a transverse craniectomy in the shape of the Greek letter pi for scaphocephaly secondary to sagittal synostosis), anterior calvarial remodeling procedures including bicoronal craniectomy with fronto-orbital advancement (FOA) and posterior cranial vault remodeling (CVR) with and without barrel stave osteotomies. These techniques are associated with significant blood loss, varying between 0.2 and 4 blood volumes, lengthy surgical times (3 to 8 hours) and hospital stays (4 to 7 days), and require often postoperative monitoring in an ICU.
B. Craniofacial syndromes
1. Clinical issues
a. Common craniofacial syndromes associated with craniosynostosis include Apert, Crouzon, Carpenter, Saethre–Chotzen, Pfeiffer, and Muenke (Table 18.1).
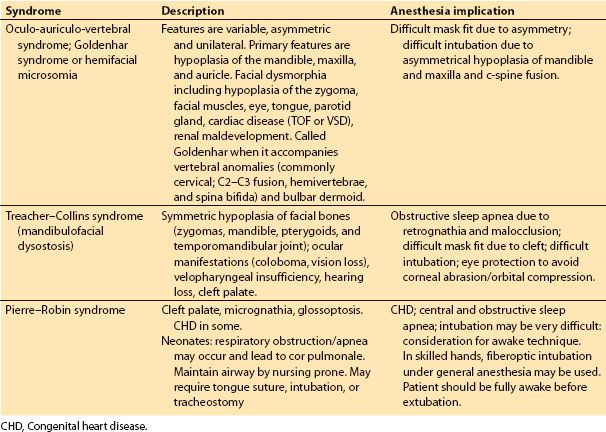
b. Common craniofacial syndromes not associated with craniosynostosis are Goldenhar/hemifacial microsomia, Treacher-Collins, and Pierre-Robin sequence (Table 18.2). However, these syndromes are associated with difficult airway management issues.
Table 18.2 Common craniofacial syndromes not associated with craniosynostosis
2. Surgical management
a. Syndromes associated with craniosynostosis. Infants may present for craniosynostosis surgery between 3 and 6 months of age or older and require surgical correction of midface/maxillary hypoplasia usually between the ages of 5 and 8 years. In adolescence, the maxillary deficiency may be corrected. In more severe cases, hypertelorism may accompany midface flattening and may require further surgical intervention.
b. Syndromes not associated with craniosynostosis. In Goldenhar/hemifacial microsomia, skeletal reconstruction involves correction of mandibular hypoplasia and soft tissue reconstruction (ear cartilage and lobe anomalies). In Treacher-Collins (mandibulofacial dysostosis) and Pierre-Robin syndrome, airway management is the primary concern, and early distraction osteogenesis (DO) to treat mandibular hypoplasia or retrognathia may avert the need for tracheostomy in severely affected neonates. Additional surgery in Treacher-Collins to correct facial bone hypoplasia is most successfully accomplished in staged procedures that address the zygomas, maxilla, and mandible along with the colobomas, macrostomia, and microtia. These reconstructions are ideally preformed between 4 and 10 years of age.
CLINICAL PEARL
Craniofacial surgery includes reconstructive surgery for craniosynostosis, cranial base tumors, encephalocele, hypertelorism, facial clefts, midface/maxillary hypoplasia, and mandibular hypoplasia. Multiple-staged procedures may be required for craniofacial reconstruction.
C. Anomalies of the face and jaw
1. Clinical issues
a. Craniofacial cleft. Craniofacial clefts are caused by failure of midline closure and lack of fusion of facial prominences. Defects involve the underlying cranial and/or facial skeletal and/or brain and/or soft tissue. Various combinations of eye, ear, and central nervous system deformities are associated with clefts. Thirty percent of patients with clefts have associated cardiac anomalies.
The most severe clefts are the midline craniofacial clefts associated with cerebrofacial dysmorphogenesis. The most severe form, holoprosencephaly, derives from the failure of the anterior portion of the neural tube to form the cerebral hemispheres, resulting in a single (holo) forebrain (prosencephaly). Infants with lobar brain morphology are mentally deficient but have a good prognosis for life expectancy and are generally repaired.
Treacher-Collins syndrome (mandibulofacial dysostosis) is characterized by eyelid coloboma, aplasia/hypoplastic zygomas and pterygoids, mandibular and midface hypoplasia, ear deformities, and poorly developed supra-orbital ridges. Intelligence is usually normal. Oculoauriculovertebral spectrum (hemifacial microsomia or Goldenhar syndrome) is the most common craniofacial malformation other than cleft lip and palate. Frontonasal dysplasia (median cleft face syndrome) is characterized as a combination of hypertelorism and median facial clefting of the nose, upper lip, premaxilla, and palate.
b. Hypertelorism. Hypertelorism is a descriptive term for an increased intraorbital distance and can lead to strabismus and abnormal binocular vision. It is caused by craniofrontonasal dysplasia, facial clefting, encephalocele, trauma, craniofacial dysostosis, and others (fibrous dysplasia, tumors, Grieg’s syndrome, and Optiz syndrome) and is often associated with one of the craniofacial dysmorphic malformations (Apert, Crouzon, Pfeiffer, and Carpenter syndromes).
Children with severe hypertelorism may not be able to develop stereoscopic vision if treatment is delayed. However, early repair may affect normal growth of the cranial skeleton. Surgery is typically delayed until 6 to 8 years of age if vision impairment is not an issue and may be staged. An intracranial approach consisting of a bifrontal craniotomy frees the orbital frame from the surrounding bone through intra- and extraorbital osteotomies.
c. Midface/maxillary hypoplasia. In Apert and Crouzon syndromes, midface hypoplasia may be treated by a Leforte III osteotomy, which involves dysjunction of the entire midface and advancement of the nose, maxilla and orbits via a bicoronal scalp incision, various osteotomies to free and advance the entire facial block with bone grafts and rigid internal fixation.
d. Mandibular hypoplasia. Various manifestations of mandibular hypoplasia are characterized by the following syndromes: Hemifacial microsomia (Goldenhar syndrome), mandibulofacial dysostosis (Treacher-Collins syndrome), and Pierre-Robin sequence. The classic syndrome of mandibular hypoplasia is Pierre-Robin sequence and is associated with acute airway obstruction, failure to thrive, and chronic hypoxia, which may lead to cerebral impairment, pulmonary hypertension, cor pulmonale, and death. Treatment depends on the following factors: (1) Extent and severity of desaturation during sleep, (2) correction of desaturation with supplemental oxygen, (3) a persistently high end-tidal carbon dioxide level, (4) failure to thrive despite nutritional and oxygen therapy.
2. Surgical management
a. Midface/maxillary hypoplasia
(1) Leforte I osteotomy corrects malocclusion due to underdevelopment of the maxilla. It involves separating the maxilla and the palate from the skull above the roots of the upper teeth through an intraoral incision, which is made horizontally across the nasal floor, anterior maxilla, and through the pterygomaxilllary junction. The maxilla is mobilized downward, advanced and stabilized with titanium screws and plates using bone grafts to fill in the spaces to insure healing and union across the bone cuts. The bone graft is frequently harvested from the iliac crest but may be harvested from the chin or lower jaw if a mandibular sagittal split osteotomy is performed at the same time.
(2) Leforte II osteotomy is used to advance the lower maxilla and entire nose forward. It is a much more involved operation than the Leforte I, as it takes longer and incurs more significant blood loss. It involves a bicoronal scalp incision combined with an intraoral incision. The osteotomy involves mobilization and advancement of the lower maxilla via osteotomies across the nasal bridge, bilaterally down the nasal bridge and through the inferior orbital rim, across the maxilla to the pterygomaxillary junction. Rigid internal fixation devices are used and defects are filled in with bone grafts.
(3) Leforte III osteotomy is used for complete underdevelopment of the midface involving the upper jaw, nose and cheek bones (as seen in Apert and Crouzon syndromes). It involves dysjunction of the entire midface and advancement of the nose, maxilla, and orbits through a bicoronal scalp incision and various osteotomies to free and advance the entire facial block. Bone grafts are used to fill in the spaces and rigid internal fixation is utilized.
Midface dysjunction, advancement, and reconstruction procedures with osteotomies can be performed simultaneously or in stages. Timing is usually not until the child is 4 or 5 years old or later but may be delayed until adolescence to allow for eruption of permanent dentation.
b. Mandibular hypoplasia. Surgical interventions depend on the severity of patency of the patient’s airway. Most infants are treated with conservative measures such as prone positioning, especially during feeding. Infants who have continued severe apnea often require surgical intervention, which traditionally consists of a tongue lip adhesion or tracheostomy. In most centers, these techniques have given way to early mandibular distraction osteogenesis.
Distraction osteogenesis (DO) is also called callotasis (stretching of the callus, as in a fracture). An osteotomy is used to fracture the bone and thereby induce a callus at which point the proximal and distal bone ends are gradually distracted and moved apart to allow new bone growth. It has the benefit of simultaneously increasing bone length and the volume of the surrounding soft tissues and has been primarily applied to treat mandibular hypoplasia. However, its use is expanding to treat other facial bone defects including midface hypoplasia (maxilla and zygoma are hypoplastic in children with severe hypoplasia as in Crouzon’s, Apert’s and hemifacial microsomia), and calvaria. External and/or internal uniplanar or multiplanar fixation devices are inserted. The three clinical phases of DO are (1) latency phase, 5 to 7 days after corticotomy and/or osteotomy when the initial fracture healing bridges cut the bony surface; (2) distraction phase, the 3 to 5-week period of active stretching when the bone is moved in three dimensions by rotating screws to increase the distraction by millimeters on a daily basis; and (3) mineralization phase, the 7 to 9-week period after distraction when the primary mineralization and strengthening of the bone occurs.
CLINICAL PEARL
Pediatric patients with craniofacial abnormalities because of their complexity, associated syndromes and co-morbidities continue to provide the anesthesiologist with some of the most challenging cases that present to the operating room.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


