CHAPTER 33
Coagulopathies
Evangelina Berrios-Colon, PharmD, MPH, BCPS, CACP • Julie Anne Billedo, PharmD, BCACP
Coagulopathies include hemorrhage, thrombosis, and embolism, and represent common clinical manifestations of hematological disease. Normally, bleeding is controlled by a fibrin clot formation, which results from the interaction of platelets, plasma proteins, and the vessel wall. The fibrin clot is ultimately dissolved through fibrinolysis. A derangement of any of these components may result in a bleeding or thrombotic disorder. In this chapter, individual disease states are examined under the broad headings of coagulation factor deficiencies, disorders of platelets, mixed disorders, acquired thrombophilias, and inherited thrombophilias.
 ANATOMY, PHYSIOLOGY, AND PATHOLOGY
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
Coagulation is initiated after blood vessels are damaged, enabling the interaction of blood with tissue factor, a protein present beneath the endothelium (Figure 33.1). Small amounts of Factor VII present in plasma bind to tissue factor, and this tissue factor–Factor VII complex activates Factor X. Activated Factor X, in the presence of Factor V, activates prothrombin (II) to thrombin (Ila), which subsequently cleaves fibrinogen to fibrin. The fibrin polymerizes into an insoluble gel. This is stabilized by the action of Factor XIII. This process constitutes the extrinsic pathway.
Coagulation is consolidated by the intrinsic pathway. Factor XI is activated (possibly by thrombin generated in the extrinsic pathway), resulting in the activation of Factor IX, which then activates Factor X in the presence of Factor VIII. Activated Factor X produces a fibrin clot, as outlined in the description of the extrinsic pathway. Decreased levels of clotting factors may be caused by defective synthesis, excessive use, circulating inhibitors of clotting factors, or excessive proteolysis by the fibrinolytic system.
The coagulation pathway is controlled by a number of endogenous anticoagulants. Protein C is a plasma protein that is vitamin K dependent. It requires activation by a complex of thrombin and thrombomodulin, an endothelial cell protein, to inhibit activated Factors V and VIII, thus inhibiting the activation of Factors IX and X, respectively. Activated protein C (APC) inhibition is catalyzed by protein S, another vitamin K–dependent plasma protein, and also requires the presence of platelet phospholipid and calcium. Antithrombin III (AT III) primarily inhibits the activity of thrombin and Factor X by binding to the factors and blocking their activity. This inhibition is greatly enhanced by heparin. Loss of function and/or decreased concentrations of these proteins result in uninhibited coagulation and hence a predisposition to spontaneous thrombosis otherwise known as a hypercoagulable state.
Fibrinolysis is a mechanism for dissolving fibrin clots. Plasmin, the activated form of plasminogen, cleaves fibrin to produce soluble fragments. Fibrinolytics, such as tissue plasminogen activator, streptokinase, and urokinase, activate plasminogen, resulting in dissolution of a fibrin clot.
 CLASSES OF BLEEDING DISORDERS
CLASSES OF BLEEDING DISORDERS
Vascular defects usually cause bleeding only into the skin and mucous membranes. Congenital causes include Osler–Weber–Rendu syndrome and Ehlers–Danlos disease. Acquired causes of vascular purpura include infections, drugs, uremia, connective tissue disorders, and dysproteinemias. Treatment is directed to the primary illness.
Inherited Coagulation Disorders
Hereditary bleeding disorders are extremely rare. The most common bleeding disorders include hemophilia A, hemophilia B, and von Willenbrand disease (vWD). The worldwide incidence of hemophilia A is 1 case per 5,000 live male births. Hemophilia B occurs at an even lower rate, with 1 case per 50,000 live male births (Soucie, Evatt, & Jackson, 1998). vWD is the most common of the three bleeding disorders across all ethnic groups, affecting 1% of the population. Up to 80% of affected patients have Type I vWD (Zimmerman & Valentino, 2013).
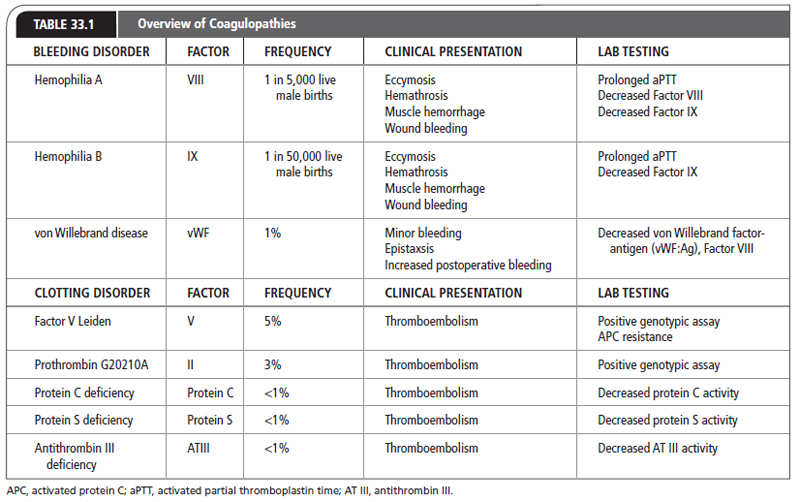
Deficiencies of any of the known coagulation factors could be present from birth. Deficiencies may be inherited or result from a spontaneous disruption in the associated coagulation factor genes. The more common deficiencies of Factors VIII and IX, as well as vWD, are discussed in this chapter (Table 33.1).
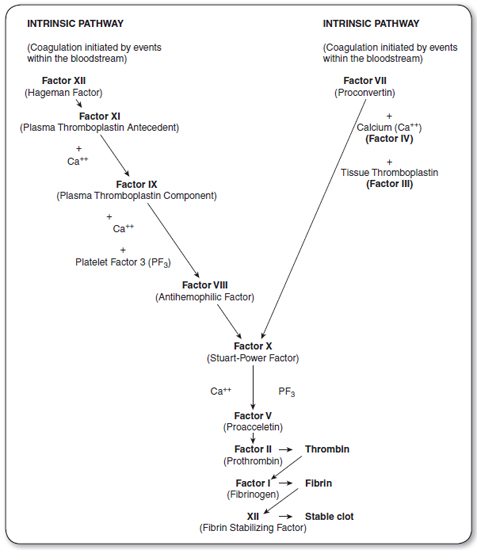
FIGURE 33.1
The coagulation cascade is initiated by the extrinsic pathway and consolidated by the intrinsic pathway. Coagulation is controlled by the inhibition of Factor VIII and Factor V by activated protein C (APC), and by antithrombin III (AT III) inhibition of Factors II and X.
HEMOPHILIA
Hemophilia is an X-linked recessive disorder associated with a congenital deficiency of Factor VIII or Factor IX. Factor VIII deficiency is known as hemophilia A and Factor IX deficiency is known as hemophilia B. Hemophilia B was first diagnosed by Steven Christmas and thus is also called Christmas disease. Hemophilia is suspected in any male who has excessive bleeding after trauma, or spontaneous bleeding into joints or soft tissues (Table 33.1). Patients with severe hemophilia (factor level: <1%) are at risk for spontaneous hemarthrosis and soft-tissue bleeding. Patients who have moderate disease (factor level: 1%–4%) or mild hemophilia (factor level: 5%–25%) are at a reduced risk of spontaneous hemorrhage, but may bleed excessively after trauma or surgery (Wagenman, Townsend, Mathew, & Crookston, 2009).
VON WILLEBRAND DISEASE
von Willebrand factor (vWF) is a plasma protein that is required for the adhesion of platelets to sites of vascular damage. In persons with von Willebrand disease, deficiency of vWF is called Type I; qualitative abnormality of vWF is called Type II. vWD is an autosomally inherited hemostatic disorder of variable severity, characterized by mucosal and cutaneous bleeding, similar to patients with platelet disorders (see Table 33.2). Patients may have a prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT).
Mixed Coagulation Disorders
Mixed coagulation disorders are a group of acquired diseases. They usually involve multiple elements of the hemostatic system and are often associated with a particular disease or clinical syndrome.
DISSEMINATED INTRAVASCULAR COAGULATION
Disseminated intravascular coagulation (DIC) can be caused by the activation of either the coagulation or fibrinolytic system, resulting in excessive bleeding or thrombosis. Conditions associated with DIC include gram-negative sepsis infections, meningococcemia, Rocky Mountain spotted fever, typhoid fever, obstetric complications (abruptio placentae, eclampsia, retained dead fetus), massive trauma, surgery, shock, and certain malignancies.
LIVER DISEASE
Coagulation disorders are common in patients with acute and chronic hepatic diseases. These may arise from malabsorption of vitamin K, decreased synthesis of clotting proteins, abnormal synthesis of clotting proteins, or decreased clearance of activated factors. Fibrin degradation products may be elevated because of poor hepatic clearance, resulting in impaired coagulation resembling DIC. Liver disease associated with hypersplenism may result in thrombocytopenia.
VITAMIN K DEFICIENCY
Factors II, VII, IX, and X are vitamin K–dependent coagulation factors that are synthesized in the liver. Vitamin K is naturally synthesized by intestinal bacteria. Vitamin K is a lipid-soluble vitamin, which is absorbed only in the presence of bile salts. Depletion of vitamin K–dependent factors may occur in patients receiving certain antibiotics; in obstructive jaundice, malabsorptive states, or hepatic parenchymal disease; and in patients receiving warfarin therapy. Patients have an elevated PT resulting in an increased international normalized ratio (INR).
Platelet Disorders
Platelet disorders include thrombocytopenia, which may be caused by diminished platelet production, enhanced platelet destruction, or sequestration of platelets (Table 33.2). Qualitative platelet disorders are more commonly acquired, but could be congenital. Congenital disorders affect platelet adhesion, aggregation, or secretion, and are rare. Acquired qualitative platelet disorders are secondary to uremia, myeloproliferative disorders, drugs, and dysproteinemias.
IMMUNE THROMBOCYTOPENIC PURPURA
Immune thrombocytopenic purpura (ITP), the autoimmune destruction of platelets, usually presents as bruising, petechiae, or bleeding. The differential diagnosis includes aplastic anemia, sepsis, DIC, acute leukemia, drug-induced thrombocytopenia, and infiltrative bone marrow disorders. The diagnosis is made by excluding other causes of thrombocytopenia, after a careful history, physical examination, and peripheral smear. An HIV test should be performed in those patients with risk factors. Bone marrow aspiration may be indicated in elderly patients or patients with atypical findings.
Causes of Thrombocytopenia |
Diminished platelet production
Marrow infiltration with tumor, fibrosis, infection
Aplastic/hypoplastic anemia
Exposure to environmental toxins (arsenic, pesticides)
Ionizing radiation
Nutritional deficiencies (B12, folate)
Viral infections
Drugs (thiazides, alcohol, myelosuppressive agents)
Paroxysmal nocturnal hemoglobinemia
Splenic sequestration
Lymphoproliferative disorders
Portal hypertension
Myeloproliferative disorders
Infections (bacterial, viral, parasitic)
Increased platelet destruction
Nonimmune
Vascular prosthesis DIC
Sepsis/infection
TTP
Immune
Drug-induced antibodies
ITP
DIC, disseminated intravascular coagulation; ITP, immune thrombocytopenic purpura; TTP, thrombotic thrombocytopenic purpura.
ITP may be acute (short term) or chronic. Acute ITP occurs primarily in children and usually lasts <6 months. Chronic ITP usually occurs in adults and lasts for >6 months, with a 3:1 ratio of females to males. It is common in patients between the ages of 20 and 50 years. ITP may also be associated with HIV infection, systemic lupus erythematosus, lymphoproliferative disorders, ulcerative colitis, and carcinoma. The thrombocytopenia is often chronic and unremitting, requiring definitive therapy.
DRUG-INDUCED THROMBOCYTOPENIA
Drug-induced thrombocytopenia is diagnosed after excluding other causes, and by noting a temporal relation between the onset of thrombocytopenia and the administration of the drug, as well as resolution upon discontinuation of the drug. Although many drugs can be implicated, alcohol, thiazide diuretics, quinine, quinidine, penicillins, gold, sulfa, and heparin are the most common. Myelosuppressive drugs used to treat malignancies and other disorders can produce thrombocytopenia by suppressing platelet production.
Heparin-induced thrombocytopenia is associated with an early nonimmune clinical syndrome and a later immune-mediated thrombocytopenia that occurs 5 to 7 days after the initiation of heparin. This type of thrombocytopenia may be associated with paradoxical thrombosis instead of bleeding.
In all cases, the offending drug should be discontinued and switched to an alternative if possible.
THROMBOTIC THROMBOCYTOPENIC PURPURA
Thrombotic thrombocytopenic purpura (TTP) is a complex clinical syndrome characterized by a formation of blood clots under the skin. Patients may present with headaches, blurred vision, seizures, profound coma, purpura, and petechiae from thrombocytopenia, jaundice, hemolytic anemia, fever, or renal dysfunction. TTP may be acquired or congenital. The causes of TTP are unknown, although this disease has been associated with certain drugs, pregnancy, malignancy, and HIV infection.
Laboratory findings include anemia, thrombocytopenia, fragmented red blood cells seen on peripheral smear, elevated blood urea nitrogen and creatinine levels, proteinuria, hematuria, and an elevated level of lactic dehydrogenase. The PT/INR and aPTT are usually normal.
 CLASSES OF THROMBOTIC DISORDERS
CLASSES OF THROMBOTIC DISORDERS
Acquired Thrombophilias
Thrombophilia refers to a tendency to have venous thromboembolisms (VTEs) and to be at risk for recurrent episodes. Acquired causes of thrombosis include malignancy, myeloproliferative disorders, use of certain drugs, surgery, trauma, prolonged immobilization, pregnancy, antiphospholipid antibodies with or without systemic lupus erythematosus, and hyperhomocysteinemia (see discussion on Virchow’s triad in Chapter 74 for additional information).
ANTIPHOSPHOLIPID ANTIBODIES
The antiphospholipid antibodies—anticardiolipin antibody, lupus anticoagulant, and antib2-glycoportein 1—are directed against different phospholipids. The lupus anticoagulant is an antibody against the phospholipid moiety of prothrombin activator complex, which interferes with and prolongs the aPTT and less commonly increases the INR. Despite the prolonged aPTT, this disorder is not associated with bleeding, but it may be associated with thrombosis. The lupus anticoagulant most highly predicts thrombosis. Antiphospholipid antibody syndrome is a term used for patients with persistent antiphospholipid antibodies who experience at least one clinical manifestation, such as a vascular thrombosis and/or pregnancy morbidity (Ruiz-Irastorza, Crowther, Branch, & Khamashta, 2010).
A high level of homocysteine, an amino acid produced from methionine metabolism, causes an increased risk of venous and arterial thrombosis when it undergoes auto-oxidation. Normal metabolism of homocysteine relies on adequate stores of folic acid and vitamins B6 and B12. Thus, hyperhomocysteinemia is not a coagulation cascade issue but a defect in metabolism and/or increase in production. Hyperhomocysteinemia can be precipitated by acquired medical conditions, including vitamin deficiencies, or by genetic defects.
Inherited Thrombophilias
Inherited thrombophilias should be considered in patients who have a VTE and who are <45 years old, have a family history of VTEs, have had recurrent spontaneous episodes of VTEs, have had thrombosis in an unusual site (e.g., mesenteric vein, cerebral vein), and/or have had recurrent fetal losses and acquired causes have been excluded. All of the inherited thrombophilias discussed here are transmitted as an autosomal dominant trait, meaning that carriers, or heterozygotes, are affected.
The prevalence of inherited thrombophilias in the general population is relatively low. The two most common thrombophilias are Factor V Leiden (FVL) and prothrombin G20210A mutation. Both of these are more prevalent in Caucasians, with a prevalence of about 5% for Factor V Leiden and 3% for prothrombin G20210A mutation. A majority of those with either disease are heterozygote carriers. Being a carrier for both Factor V Leiden and prothrombin mutation is rare, with a prevalence of about 0.1% (Lijfering et al., 2010). Deficiencies of the natural anticoagulants have a prevalence of <1% in the general population. The least common is antithrombin deficiency (Seligsohn & Lubetsky, 2001). People can be carriers of multiple thrombophilia disorders, each of which further increases a person’s lifetime risk for a VTE.
FACTOR V LEIDEN
A mutation of a single DNA base pair found on the Factor V gene causes the production of defective Factor V proteins. This single base change, where guanine is substituted with adenine, leads to the replacement of arginine by guanine at position 506, creating Factor V Leiden. These defective Factor V proteins resist APC. In other words, this mutation causes resistance of Factor V to the endogenous anticoagulation effects of protein C and causes an imbalance of the clotting cascade favoring thrombosis. The Factor V Leiden mutation was discovered by researches in Leiden, Netherlands, and is the most commonly diagnosed thrombophilia. Heterozygote carriers of Factor V Leiden are at a five-fold increased risk for a first-time VTE, and homozygote carriers are at an 18-fold increased risk, compared to the general population (Lijfering et al., 2010).
PROTHROMBIN G20210A MUTATION
Prothrombin G20210A mutation is a single base pair mutation on the 3′-untranslated region of the prothrombin gene. At position 20210 in this region, guanine is substituted with adenine. This substitution leads to increased production of prothrombin and thus thrombin. Both heterozygote and homozygote carriers have elevated levels of prothrombin. Heterozygote carriers of prothrombin G20210A mutation are at a threefold increased risk for a first-time VTE compared to the general population (Lijfering et al., 2010).
ANTITHROMBIN DEFICIENCY
Lack of antithrombin activity enables the coagulation cascade to continue uninterrupted. Antithrombin deficiency results from a quantitative or qualitative defect and is divided into two different types depending on its origin. Type I antithrombin deficiency is quantitative and occurs when production of normally functioning antithrombin is decreased. Type II antithrombin deficiency is qualitative and occurs when dysfunctional antithrombin molecules are produced. Certain conditions can induce a state of antithrombin deficiency. Such conditions associated with acquired antithrombin deficiency include heparin therapy, liver and/or renal disease, and DIC (Seligsohn & Lubetsky, 2001).
PROTEIN C DEFICIENCY
Protein C deficiency results from either a quantitative or a qualitative defect and is divided into two different types depending on its origin. Type I protein C deficiency is quantitative and occurs when production of normally functioning protein C molecule is decreased. Type II is qualitative and occurs when defective protein C molecules are produced, leading to low activity of protein C. Type I defect predominates in protein C deficiency. Certain conditions can cause protein C deficiency and/or induce further deficiency. Such conditions associated with acquired protein C deficiency include liver disease, vitamin K deficiency, use of vitamin K antagonists (VKAs), and DIC (Seligsohn & Lubetsky, 2001).
PROTEIN S DEFICIENCY
As with the other deficiencies, protein S deficiency results from a quantitative or qualitative defect; Type I and Type II, respectively. However, protein S deficiency has another category, Type III. Type III protein S deficiency is due to decreased amounts of free protein S molecules. Approximately 60% of protein S is bound to C4b-binding protein, an acute phase reactant, and the remaining free portion of protein S has the ability to be the cofactor that helps activate protein C. Free protein S can be decreased due to increased C4b-binding proteins or due to a divergence between protein S and C4b-binding protein to complex. Types I and III predominate in protein S deficiency. As with the other deficiencies, certain situations can cause protein S deficiency. Situations associated with acquired protein S deficiency include pregnancy, oral contraceptive therapy, inflammatory conditions, liver disease, vitamin K deficiency, use of VKAs, and DIC (Marlar & Gausman, 2011; Seligsohn & Lubetsky, 2001).
 HISTORY AND PHYSICAL EXAMINATION
HISTORY AND PHYSICAL EXAMINATION
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


