KEY POINTS
1. Previous concepts of independent intrinsic and extrinsic plasma coagulation pathways, a cell-free final common pathway, and platelet clotting have given way to an integrated concept of cell-based coagulation occurring on the platelet surface as depicted in Figure 19.2.
2. In the absence of a history suggesting a preoperative bleeding disorder (e.g., von Willebrand’s Disease, warfarin therapy), routine hemostatic screening for patients undergoing cardiac surgery is not cost-effective at predicting excessive perioperative bleeding.
3. Unfractionated heparin (UFH) is a hydrophilic macromolecular glycosaminoglycan of varying chain lengths which anticoagulates principally by potentiating antithrombin III (AT III)-induced inactivation of factors IIa (thrombin) and Xa.
4. Intravenously administered heparin typically peaks within 1 min, redistributes only slightly, and has a dose-related half-life that reaches approximately 2 hrs in the large doses used for cardiopulmonary bypass (CPB). Heparin bolus administration decreases systemic vascular resistance by 10% to 20%.
5. The usual initial heparin dose is 300 to 350 units/kg to achieve an Activated Clotting Time (ACT) exceeding 400 s for safe initiation and maintenance of CPB. Heparin 5,000 to 10,000 units should be added to CPB priming solution.
6. Heparin is most often monitored using ACT, but ACT precision is suboptimal at the heparin concentrations required for CPB, and it is also subject to prolongation from hemodilution and hypothermia. As a result, some practitioners choose to monitor and maintain a target whole blood heparin concentration (typically 3 to 4 units/mL) in addition to exceeding a target ACT during CPB.
7. Resistance to heparin-induced anticoagulation has a variety of causes, but therapy with either AT III concentrates or fresh frozen plasma (FFP) nearly always resolves it regardless of its cause.
8. Heparin-induced thrombocytopenia (HIT) produces a severe procoagulant state that may occur after 5 or more continuous days of heparin administration. Diagnosis requires the combination of an appropriate clinical context and a complex variety of laboratory tests.
9. Documented presence of HIT in a patient who must urgently undergo cardiac surgery requiring CPB is possibly best managed by using bivalirudin for anticoagulation.
10. Protamine neutralizes heparin stoichiometrically as a result of a strong cation (protamine)-to-anion (heparin) interaction.
11. A variety of protamine dosing methods are used clinically. Most often 60 to 80 mg of protamine per 100 units of heparin administered prior to CPB (including heparin placed in the CPB priming solution if residual CPB volume is to be reinfused unaltered by hemoconcentration or cell washing) suffices to neutralize heparin. Excessive protamine administration impairs blood clotting.
12. In order to avoid hypotension, protamine should be administered slowly, ideally by a continuous intravenous infusion over 5 to 10 min.
13. Protamine can induce severe anaphylactic or anaphylactoid reactions.
14. Post-CPB clotting abnormalities are best managed using a systematic approach. Post-CPB bleeding algorithms reduce bleeding and transfusion.

IN ESSENCE, CPB CREATES a blood “detour” to permit surgery on the heart. This detour must route the blood through an artificial heart and lung while maintaining its fluidity. Historically, fluidity represented the final frontier in the development of cardiac surgery because effective mechanisms for blood gas exchange and for propelling the blood had been established more than a decade before surmounting the fluidity challenge. The challenge was to find a therapeutic approach that would inhibit blood’s natural propensity to clot when it contacts foreign surfaces. Since the restoration of normal coagulation was desirable at the end of the surgical procedure, this clotting inhibition needed to be reversible, like turning a spigot on and off. The long-awaited solution was monumental: anticoagulation with heparin followed by neutralization with protamine. This fundamental approach to establishing and reversing blood fluidity remains unchanged after almost 50 yrs, although much fine-tuning has occurred. This chapter reviews anticoagulation and the restoration of coagulation in the patients undergoing CPB.
I. Physiology of Coagulation
A. Mechanisms of hemostasis
1. Plasma coagulation. Figure 19.1 depicts the plasma coagulation pathway. Blood contact with foreign surfaces classically was thought to activate the intrinsic pathway, whereas vascular injury or disruption was thought to activate the extrinsic pathway. These definitions seem counterintuitive because vascular disruption should be intrinsic and foreign bodies extrinsic, but logic has held little sway in coagulation pathway nomenclature. Thankfully, distinctions between the intrinsic and extrinsic pathways have become less important because both the activants and the pathways overlap (e.g., connection between VIIa and IXa). The coagulation factor numbering system was defined in order of discovery rather than use, which explains the illogic numerical progression through the pathways [1].
Figure 19.1 Schematic representation of the hemostatic system depicting the vascular, platelet, and coagulation components. F, factor; HMWK, high-molecular weight kininogen; vWF, von Willebrand’s factor; Ca++; ionized calcium; VIII:C, factor VIII coagulation component; TxA2, thromboxane A2; ADP, adenosine diphosphate.
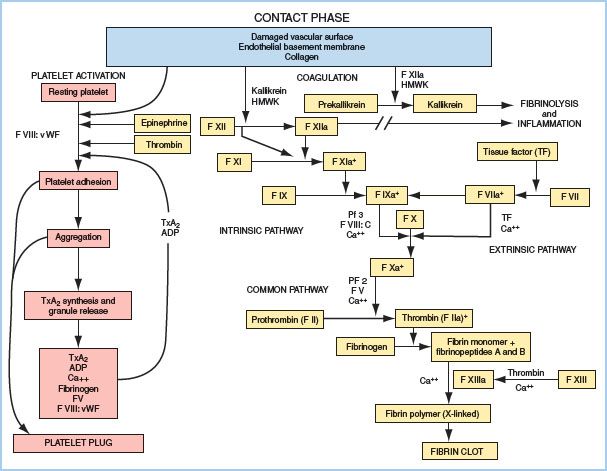
a. Intrinsic pathway. Contact activation involves binding of factor XII to negatively charged surfaces, which leads to the common pathway through factors XI, IX, platelet factor 3, cofactor VIII, and calcium. Kallikrein is also formed in this reaction and serves as a positive feedback mechanism and as an initiator of fibrinolysis (a negative feedback mechanism) and inflammation. For cardiac surgery, this pathway’s clinical importance lies more in the access it provides for heparin monitoring and neutralization than in its role in normal hemostasis.
b. Extrinsic pathway. Tissue factor (TF) initiates the extrinsic pathway, which proceeds to activate factor IX and rapidly stimulates the common pathway with the aid of factor VII and calcium.
c. Common pathway. Beginning with the assisted activation of factor X, this pathway proceeds to convert prothrombin (factor II) to thrombin and fibrinogen (factor I) to fibrin monomer, which initiates the actual substance of the clot. Fibrin monomer then cross-links to form a more stable clot with the aid of calcium and factor XIII. Rather than thinking of the common pathway as the result of activation of two independent paths, the concept of cell-based coagulation has been adapted to better explain the hemostatic mechanisms that occur simultaneously in the body (Fig. 19.2). When tissue injury occurs, TF is expressed on the surface of TF-bearing cells. Presentation of TF to its ligand factor VII causes the activation of factors IX and X on the TF-bearing cell. This is called “Initiation.” The activation of factor X causes thrombin formation which then incites further protease activation. These reactions occur on the phospholipid surface of the platelet and are called “Amplification.” The final stage of clot formation is known as “Propagation.”
Figure 19.2 Cell-based model of hemostasis. Cellular hemostasis is thought to occur in three stages: Initiation, amplification, and propagation. The initiation stage (1) takes place on TF-bearing cells (cells such as monocytes that can bind TF and present it to a ligand), which come into play when endothelial injury occurs and TF is exposed. The initiation stage is characterized by presentation of TF to its ligand factor VII and the subsequent activation of factors IX and X on the TF-bearing cell. The activation of factor X to Xa causes thrombin production and activation. Once generated, thrombin feeds back to activate factors VIII, V, and platelets. The amplification stage (2) then occurs on the surface of the activated platelet, which exposes surface phospholipids that act as receptors for the activated factors VIIIa and IXa. The platelet surface allows for further thrombin formation and hence the amplification of coagulation. Continued generation and activation of thrombin causes further positive feedback mechanisms (3) to occur that ultimately ensure the formation of a stable clot, including cleavage of fibrinogen to fibrin, release and activation of factor XIII for fibrin cross-linkage, and the release of a thrombin-activatable fibrinolysis inhibitor.
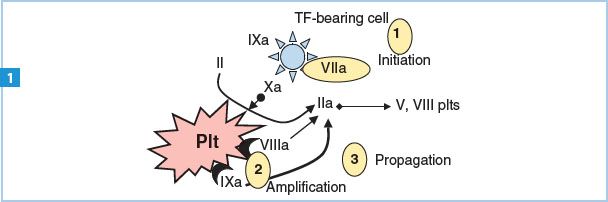
Activated factor X also initiates platelet-surface clotting activity as depicted in Figure 19.2. This platelet-surface activity greatly speeds the overall formation of clot and should be considered as a vital component of the clotting cascade.
d. Thrombin is the most important enzyme in the pathway because (in addition to activating fibrinogen) it
(1) Provides positive feedback by activating cofactors V and VIII
(2) Accelerates cross-linking of fibrinogen by activating factor XIII
(3) Strongly stimulates platelet adhesion and aggregation
(4) Facilitates clot resorption by releasing tissue plasminogen activator (tPA) from endothelial cells
(5) Activates protein C, which provides negative feedback by inactivating factors Va and VIIIa
2. Platelet activation. As shown in Figure 19.1, a variety of stimuli initiate platelet activation, and thrombin is an especially potent one. This sets off a cascade of events that initiates platelet adhesion to endogenous or extracorporeal surfaces, followed by platelet aggregation and formation of a primary platelet plug. Fibrin clots and platelet plugs form simultaneously and mesh together, yielding a product more tenacious and difficult to dissolve than either alone. Whereas formerly plasma-based and platelet-based clotting were often thought to represent independent pathways, more recent accounts show that the plasma coagulation pathway evolves mainly on the platelet surface, such that the plasma and platelet clotting processes are more interdependent than independent.
a. Von Willebrand factor (vWF) is an important ligand for platelet adhesion at low shear stress, and for aggregation at higher shear rates. Fibrinogen is the major ligand for platelet aggregation.
b. Products released from platelet storage granules (adenosine diphosphate [ADP], epinephrine, calcium, thromboxane A2, factor V, and vWF) serve to perpetuate platelet activation and the plasma coagulation cascade.
3. Nature’s system of checks and balances demands counterbalancing forces to discourage runaway clot formation and to dissolve clots. These counterbalances include the following:
a. Proteins C and S, which inactivate factors Va and VIIIa
b. ATIII, antithrombin, or AT, which inhibits thrombin as well as factors XIa, IXa, XIIa, and Xa
c. TF inhibitor, which inhibits the initiation of the extrinsic pathway
d. tPA, which is released from endothelium and converts plasminogen to plasmin, which in turn breaks down fibrin. Plasminogen activator inhibitor 1 in turn inhibits tPA to prevent uncontrolled fibrinolysis.
B. Tests of hemostatic function
1. Table 19.1 lists commonly used tests of hemostatic function [1]. These tests may be used to detect hemostatic abnormalities preoperatively or after CPB. With the exception of the ACT, typically they are not used during CPB except under extenuating circumstances, because most of them will be abnormal as a result of hemodilution, anticoagulation, and sometimes hypothermia.
1
a. Most studies suggest that routine preoperative hemostatic screening is not helpful in predicting patients who will bleed excessively during surgery. If the patient’s clinical history (e.g., nosebleeds; prolonged bleeding with small cuts, dental work, or surgery; easy bruising; strong family history of pathologic bleeding) suggests the need for hemostatic screening, selective use of these and other tests is appropriate. Similarly, when the patient is taking medications that alter hemostatic function, specific hemostatic function tests may be indicated. Examples include the following:
Table 19.1 Common clinical tests of hemostatic function
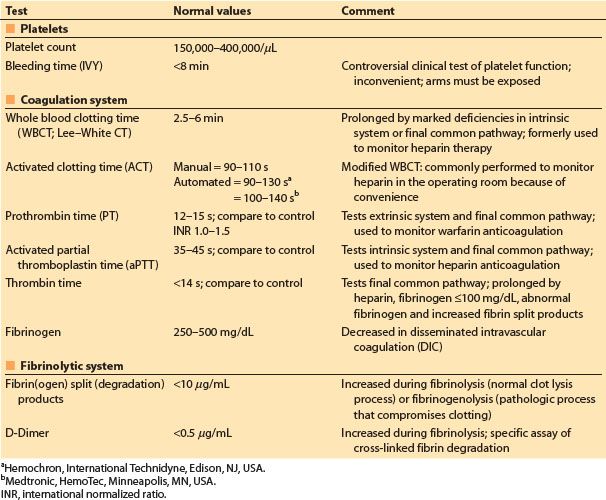
(1) Heparin: Activated partial thromboplastin time (aPTT) or ACT
(2) Low-molecular-weight heparin (LMWH), including the pentasaccharide fondaparinux: No test or anti-Xa plasma activity
(3) Warfarin: Prothrombin time (PT) and/or international normalized ratio (INR)
(4) Platelet inhibitors including aspirin: No testing, bleeding time, or specific platelet function tests. Preliminary data suggests that specific platelet function tests in patients taking thienopyridine agents may correlate with postoperative bleeding risk.
II. Heparin anticoagulation
A. Heparin pharmacology [2]
Structure. As drugs go, UFH might be described as impure. Heparin resides physiologically in mast cells, and it is commercially derived most often from the lungs of cattle (bovine lung heparin) or the intestines of pigs (porcine mucosal heparin). Commercial preparations used for CPB typically include a range of molecular weights from 3,000 to 40,000 Da, with a mean molecular weight of approximately 15,000 Da. Each molecule is a heavily sulfated glycosaminoglycan polymer, so heparin is a strong biologic acid that is negatively charged at physiologic pH.
a. Porcine mucosal and bovine lung heparin both are satisfactory for CPB and both have been widely used.
3
1. Action. A specific pentasaccharide sequence that binds to ATIII is present on approximately 30% of heparin molecules. This binding potentiates the action of ATIII more than 1,000-fold, thereby allowing heparin to inhibit thrombin and factor Xa most importantly, but also factors IXa, XIa, and XIIa.
a. Inhibition of thrombin requires simultaneous binding of heparin to both ATIII and thrombin, whereas inhibition of factor Xa requires only that heparin binds to ATIII. The former reaction limits thrombin inhibition to longer saccharide chains (18 or more saccharide units); hence, shorter chains can selectively inhibit Xa. This is the primary principle underlying therapy with LMWH and with the “ultimate” LMWH fondaparinux, which contains just the critical pentasaccharide sequence needed for binding ATIII, hence it induces virtually exclusive Xa inhibition.
(1) Because thrombin inhibition appears pivotal for CPB anticoagulation and also because LMWH and heparinoids have a long half-life and are poorly neutralized by protamine, LMWH (including fondaparinux) is inadvisable as a CPB anticoagulant. Fondaparinux lasts even longer than traditional LMWHs and is even less neutralized by protamine.
b. Heparin binds and activates cofactor II, a non–ATIII-dependent thrombin inhibitor. This may explain why heparin-induced anticoagulation can be effective even in the presence of marked ATIII deficiency, although the primary mechanism of anticoagulant action is ATIII inhibition.
2. Potency. Heparin potency is tested by measuring the anticoagulation effect in animal plasma. The United States Pharmacopoeia (USP) defines 1 unit of activity as the amount of heparin that maintains the fluidity of 1 mL of citrated sheep plasma for 1 hr after recalcification.
a. Heparin dosing is best recorded in USP units, because commercial preparations vary in the number of USP units per milligram. The most common concentration is 100 units/mg (1,000 units/mL) [3].
4
3. Pharmacokinetics. After central venous administration, heparin’s effect peaks within 1 min, and there is a small rapid redistribution that most often is clinically insignificant [4,5].
a. Heparin’s large molecular size and its polarity restrict its distribution mainly to the intravascular space and endothelial cells.
b. The onset of CPB increases circulating blood volume by approximately 1,000 to 1,500 mL; hence, plasma heparin concentration drops proportionately with the onset of CPB unless heparin is added to the CPB priming solution.
c. Heparin is eliminated by the kidneys or by metabolism in the reticuloendothelial system.
d. Elimination half-life has been determined only by bioassay, that is, by the time course of clotting time prolongation. By this standard, heparin’s elimination time is dose dependent [6]. At lower doses, such as 100 to 150 USP units/kg, elimination half-time is approximately 1 hr. At CPB doses of 300 to 400 USP units/kg, elimination half-time is 2 or more hours; hence, clinically significant anticoagulation might persist for 4 to 6 hrs in the absence of neutralization by protamine. Hypothermia and probably CPB itself prolong elimination.
4. Side effects. Heparin’s actions on the hemostatic system extend beyond its primary anticoagulant mechanism to include activation of tPA, platelet activation, and enhancement of TF pathway inhibitor.
a. Lipoprotein lipase activation influences plasma lipid concentrations, which indirectly affects the plasma concentrations of lipid-soluble drugs.
b. Heparin boluses decrease systemic vascular resistance. Typically this effect is small (10% to 20%), but rarely it can be more impressive and may merit treatment with a vasopressor or calcium chloride.
c. Anaphylaxis rarely occurs.
d. HIT is covered elsewhere in this chapter.
5
B. Dosing and monitoring
1. Dosing. The most common initial dose for CPB is 300 to 350 USP U/kg. Some centers choose an initial dose of 400 U/kg or base the initial dose on a bedside ex vivo heparin dose-response titration.
a. Since heparin distributes primarily into the plasma compartment, increasing the dose with increasing body weight assumes that plasma volume increases in direct proportion to body weight. This is not the case, because fat does not increase blood volume in proportion to weight. Consequently, there is seldom reason to exceed an initial dose of 35,000 to 40,000 units, even in patients weighing more than 100 kg, as lean body mass tends to peak at 90 kg for females and 110 kg for males.
b. Heparin dosing for coronary revascularization procedures performed without CPB is controversial. Published doses range from 100 to 300 U/kg, but most centers use 100 to 150 units/kg and set minimum acceptable ACT values at 200 to 300 s.
c. The CPB priming solution should contain heparin at approximately the same concentration as that of the patient’s bloodstream at the onset of CPB. Since this most often would be 3 to 4 U/mL, a priming volume of 1,500 mL should contain at least 5,000 units of heparin. CPB priming solutions commonly contain 5,000 to 10,000 units of heparin.
d. Supplemental heparin doses typically are guided by monitoring of anticoagulation.
2. Monitoring. Until the late 1970s, heparin dosing was guided by experiential practices and varied a great deal from hospital to hospital. Using ACT, a variation on the Lee–White clotting time, Bull et al. [7] identified rather staggering variations in the approach to heparin dosing and in both the initial anticoagulant response and the time course of anticoagulation in response to a fixed dose of heparin. This landmark work rapidly led to the realization that the anticoagulant response to heparin should be monitored, although a few centers continue to dose heparin empirically.
3. Approaches to anticoagulation monitoring for CPB. The ACT is the most widely used test, although some centers monitor blood heparin concentration as well.
a. ACT uses an activant such as celite or kaolin to activate clotting, then measures the clotting time in a test tube. Heparin prolongs ACT with a roughly linear dose-response pattern (Fig. 19.3). Normal ACT depends upon such factors as the specific activant and device, prewarming (vs. room temperature) of test tubes, and operator technique, but generally falls between 110 and 140 s.
Figure 19.3 Graph of a heparin (and protamine) dosing algorithm. In the graph, the control ACT is shown as point A and the ACT resulting from an initial heparin bolus of 200 units/kg is shown in point B. The line connecting A and B then is extrapolated and a desired ACT is selected. Point C represents the intersection between this line and a target ACT of 400 s, theoretically requiring an additional heparin dose represented by the difference between points C and B on the horizontal axis (arrow C). Similarly, to achieve an ACT of 480 s (higher horizontal dotted line intersecting the ACT versus heparin dose line at point D), one would administer the additional heparin dose represented by arrow D. To estimate heparin concentration and calculate protamine dose at the time of heparin neutralization, the most recently measured ACT value is plotted on the dose-response line (point E in the example). The whole blood heparin concentration present theoretically is represented by the difference between point E and point A on the horizontal axis (arrow E). The protamine dose required to neutralize the remaining heparin then may be calculated. Protamine 1.0 mg/kg is administered for every 100 units/kg of heparin present. (Modified from Bull BS, Huse WM, Brauer FS, et al. Heparin therapy during extracorporeal circulation: II. The use of a dose-response curve to individualize heparin and protamine dosage. J Thorac Cardiovasc Surg. 1975;69:686; and Gravlee GP. Anticoagulation for cardiopulmonary bypass. In: Gravlee GP, Davis RF, Kurusz M, et al., eds. Cardiopulmonary Bypass: Principles and Practice, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2000:435–472, with permission.)
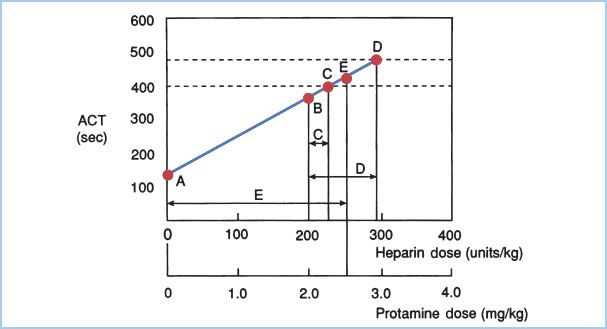
Although originally described as a manual test, most centers use one of the two automated approaches to ACT (International Technidyne, Edison, NJ, U.S.A., or Medtronic HemoTec, Fridley, MN, U.S.A.). These two automated approaches yield slightly different values both at baseline and with anticoagulation because of different activators and endpoint detection techniques.
ACT is prolonged by hypothermia and hemodilution; hence, conditions often imposed by CPB alter the ACT-heparin dose-response relationship [8]. Some see this as risking underanticoagulation, although hemodilution and hypothermia legitimately enhance anticoagulation. Overreliance on hypothermic enhancement of ACT prolongation risks underanticoagulation upon rewarming. Also, at temperatures below 25°C, ACT prolongation becomes so profound that alternative tests such as whole blood heparin concentration measurement may be advisable.
The target ACT level for CPB is controversial. There are studies supporting the safety of ACT values as low as 300 s, inside and outside of the context of heparin-coated surfaces, yet most centers accept only values exceeding 400 to 480 s. In addition, different devices and tests yield different dose-response relationships for heparin concentration versus ACT, as well as different sensitivities to hypothermia and hemodilution.
6
(1) As a clotting test, ACT is somewhat crude and may vary as much as 10% on repeated testing at heparin concentrations used for CPB [9], so it seems reasonable to build in a safety margin by accepting 400 s as a minimum safe threshold for sustained CPB.
(2) Aprotinin, which is now used rarely in clinical practice, complicates the use of ACT monitoring, as a result of marked prolongation of celite ACTs in the presence of heparin and aprotinin. This may represent enhanced anticoagulation to some degree, but in the presence of aprotinin it is probably wise to titrate heparin to a celite ACT level exceeding 750 s or to use a kaolin ACT instead. Kaolin ACT minimum levels do not have to be adjusted in the presence of aprotinin.
b. Whole blood heparin concentrations can be measured during CPB. The most commonly used technique is automated protamine titration (Medtronic HemoTec, Fridley, MN). Advocates of this monitoring technique argue that CPB-induced distortion of the ACT-heparin dose-response relationship mandates maintenance of the heparin concentration originally needed before CPB to achieve the target ACT level [10]. Heparin dosing based upon concentration alone substantially increases the amount of heparin given during CPB, which enhances suppression of thrombin formation. This benefit may accrue at the expense of heparin rebound, if not monitored, and more profound platelet activation that may aggravate and prolong platelet dysfunction after CPB. The distortion of the ACT-heparin sensitivity relationship can be partly overcome using plasma-modified ACT testing [11] or by maximal activation of the ACT test sample, as is done in a Thromboelastograph (TEG) modification of the ACT [12]. Whole blood point-of-care measurement of heparin concentration can also be performed with the HepTest POC-Hi (Americana Diagnostica, Stamford, CT). This test correlates well with heparin concentrations during CPB and more closely approximates the trend in plasma anti-Xa levels than does the standard ACT [13].
(1) Whole blood heparin concentrations of 3.0 to 4.0 U/mL most often are sufficient for CPB. Plasma heparin concentrations (typically anti-Xa concentrations) are higher because circulating heparin resides in the plasma compartment.
(2) Patients can vary widely in their sensitivity to heparin-induced anticoagulation; therefore, isolated use of heparin concentration could lead to dangerous underanticoagulation. If this technique is chosen, simultaneous use of ACT or another clotting time is strongly advised.
(3) Heparin concentration monitoring may be a useful adjunct to ACT monitoring both in the presence of aprotinin and during periods of hypothermia below 25°C.
(4) Heparin concentration monitoring may be advantageous in selecting a protamine dose, because the dose will be chosen in relation to approximate actual blood heparin concentration. The weakness of this technique is its dependence on a calculated blood volume determination at a time when blood volume may vary substantially.
c. Other monitors of anticoagulation. Neither ACT nor heparin concentration is perfect, so other tests have been evaluated or are under investigation. The aPTT and traditional thrombin time are typically so sensitive to heparin that those tests are not useful at the heparin concentrations needed for CPB. The high-dose thrombin time (HiTT, International Technidyne) offers a linear dose-response in the usual heparin concentration range used for CPB. The heparin management test (HMT, Helena, Beaumont, TX) offers another platform for ACT monitoring and can be used to monitor heparin at high (cardiac surgery) and low (vascular surgery) concentration ranges. Clotting times have also been successfully monitored using viscoelastic tests—the Sonoclot (SkACT, Sienco, Arvada, CO) [14] and the TEG.
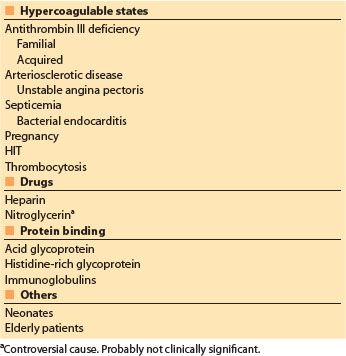
C. Heparin resistance. Heparin resistance is loosely defined as the need for greater than expected heparin doses to achieve the target ACT for CPB. As noted earlier, ACT prolongation in response to heparin varies greatly. A number of factors that may decrease the ACT response to heparin are listed in Table 19.2 [2]. ATIII deficiency is cited most often for heparin resistance, but overall the correlation between ATIII concentrations and anticoagulant response to a bolus of heparin has been weak and inconsistent, perhaps because heparin resistance often is multifactorial.
Clinical approach. Most often heparin resistance can be managed by simply giving more heparin. It is reasonable to consider administering supplemental ATIII if more than 600 USP units/kg has been required to reach the target ACT level. In some circumstances (e.g., urgent need to initiate CPB, minimal ACT prolongation after 300 U/kg), a lower intervention threshold seems reasonable. Similarly, if maintaining the target ACT during CPB requires administering more than 100 USP units/kg per 30 min of CPB, supplemental ATIII seems reasonable.
7
a. ATIII can be provided in the form of FFP, liquid plasma, and ATIII concentrates (human or recombinant).
(1) FFP or liquid plasma dose typically is 2 to 4 units in adults.
(2) For ATIII concentrates, an initial dose of 500–1,000 units is usually sufficient.
1. Prediction of heparin resistance. Medtronic HemoTec’s HMS Plus Hemostasis Management System and International Technidyne’s Hemochron RxDx system each provide an in vitro opportunity to titrate the patient’s whole blood to predetermined heparin concentrations and hence to predict the initial heparin dose required to achieve a particular target ACT. Some centers use one of these devices to predict heparin resistance prior to heparin administration. This approach allows “customization” of initial heparin dosing as well as advance preparation for anticipated heparin resistance, for example, ordering ATIII concentrate or FFP.
D. Heparin-induced thrombocytopenia (HIT)
1. Benign Thrombocytopenia from heparin’s proaggregatory effect on platelets develops in 5% to 28% of patients.
a. This mild decrease in platelet count typically occurs within 1 to 2 days of heparin administration without thrombosis or immune response. This response was formerly called HIT type I, and it is not considered pathologic. However, thrombocytopenia occurring within 1 to 2 days of heparin administration can be pathologic immune-mediated HIT if the patient’s plasma retains heparin antibodies from a previous exposure. This latter situation is most likely to occur in a patient who has experienced clinical HIT within the past several weeks.
b. HIT is a severe condition that most often occurs after 5 continuous days of heparin administration (average onset time, 9 days), and is immune mediated. Antibody binding to the complex formed between heparin and platelet factor 4 (PF4) is responsible for the syndrome. Antibody binding to platelets and endothelial cells causes platelet activation, endothelial injury, and complement activation. The platelet clots formed are referred to, in lay terms, as “white clots.” This syndrome is highly morbid and can be fatal as a result of thromboembolic phenomena.
8
(1) Among patients who develop HIT II, the incidence of thrombosis approximates 20%, the mortality of which can be as high as 40%.
(2) Diagnosis: thrombocytopenia (usually defined as platelet count <100,000/uL, but this is complicated for several days by post-CPB hemodilution), demonstration of heparin/PF4 antibody plus (ideally) documentation of heparin-induced aggregation of platelets.
(a) Heparin-induced serotonin release assay: a functional test, often considered the gold standard.
(b) Heparin-induced platelet activation assay (HIPAA): a functional test, may be nonspecific.
Table 19.2 Potential causes of heparin resistance
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


