 Hemostasis is a dynamic process. Once bleeding occurs, clot formation and degradation (fibrinolysis) are also initiated.
Hemostasis is a dynamic process. Once bleeding occurs, clot formation and degradation (fibrinolysis) are also initiated. Coagulation is an integral part of inflammation, and inflammation may lead to microvascular thrombosis.
Coagulation is an integral part of inflammation, and inflammation may lead to microvascular thrombosis. A consumptive coagulopathy (DIC) should always be considered in a patient with diffuse or generalized bleeding; however, liver disease and vitamin K deficiency are more common.
A consumptive coagulopathy (DIC) should always be considered in a patient with diffuse or generalized bleeding; however, liver disease and vitamin K deficiency are more common. Localized bleeding in a trauma or postoperative patient should prompt notification of surgical staff and search for an anatomic source of the bleeding.
Localized bleeding in a trauma or postoperative patient should prompt notification of surgical staff and search for an anatomic source of the bleeding. Thrombotic events are being recognized with increased frequency in the PICU. Only 60% of children with documented thromboses will have an identifiable hematologic abnormality.
Thrombotic events are being recognized with increased frequency in the PICU. Only 60% of children with documented thromboses will have an identifiable hematologic abnormality. Recombinant factor VIIa has been used to control refractory hemorrhage, but its use has not yet been shown to improve outcome and consequently may not justify accompanying risks.
Recombinant factor VIIa has been used to control refractory hemorrhage, but its use has not yet been shown to improve outcome and consequently may not justify accompanying risks. Neither the prothrombin time nor the International Normalized Ratio (INR) reflect bleeding risk accurately in patients with liver disease.
Neither the prothrombin time nor the International Normalized Ratio (INR) reflect bleeding risk accurately in patients with liver disease. The presence of a central venous catheter is the most commonly identified thrombotic risk factor in critically ill children.
The presence of a central venous catheter is the most commonly identified thrombotic risk factor in critically ill children. occur simultaneously, and it minimizes an identification of the contributions inflammation, platelets, and the endothelium make in the overall process. Central to this broader understanding is the realization of the primacy of Factor VII (F.VII) and TF. After damage to vascular endothelium, blood comes in contact with TF expressed on fibroblasts and leukocytes. TF triggers both the cellular component of hemostasis (by activating platelets to form a primary platelet plug) and the soluble (protein) component by binding with activated F.VII. The TF-F.VIIa complex unleashes a cascade of protein reactions that ultimately result in the production of fibrin strands, which reinforce the platelet plug.
occur simultaneously, and it minimizes an identification of the contributions inflammation, platelets, and the endothelium make in the overall process. Central to this broader understanding is the realization of the primacy of Factor VII (F.VII) and TF. After damage to vascular endothelium, blood comes in contact with TF expressed on fibroblasts and leukocytes. TF triggers both the cellular component of hemostasis (by activating platelets to form a primary platelet plug) and the soluble (protein) component by binding with activated F.VII. The TF-F.VIIa complex unleashes a cascade of protein reactions that ultimately result in the production of fibrin strands, which reinforce the platelet plug.free-flowing blood, but rather on surfaces. Platelets, endothelial cells, the subendothelial matrix, and biologic polymers (e.g., catheters, grafts, stents, etc.) provide the surfaces for clot formation.
TABLE 118.1 OVERVIEW OF COAGULATION DISORDERS | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
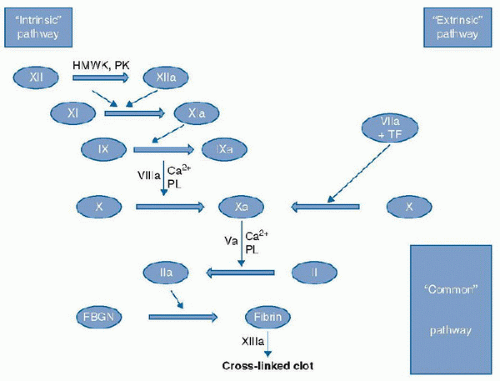 FIGURE 118.1. “Classical” coagulation cascade. Serial activation of serine proteases from zymogen to active form resulting in fibrin clot formation. Those elements in RED represent cofactor for enzymatic steps. HMWK, high-molecular-weight kininogen; PK, prekallekrein; TF, tissue factor; PL, phospholipid; Ca, calcium; Clotting factors: XII, factor XII; XIIa, activated factor XII; XI, factor XI; XIa, activated factor XI; IX, factor IX; IXa: activated factor IX; VIIIa, activated factor VIII; VIIa, activated Factor VII; X, factor X; Xa, activated factor Xa; Va, activated factor V; II, prothrombin; IIa, thrombin; FBGN, fibrinogen; XIIIa, activated factor XIII. |
 host defense. Disseminated intravascular coagulation (DIC) illustrates the link between coagulation and inflammation. In DIC, coagulation pathways are activated, natural inhibitory pathways of coagulation are dysfunctional, and the fibrinolytic system is dysregulated. All of these are direct or indirect consequences of the inflammatory response. The natural inhibitory pathways of coagulation are of particular interest in this intersection of coagulation and inflammation, as potential therapies have been based around these biologic processes (6,7). Coagulation may be initiated in the flowing blood, on the endothelial surface, at endothelial lesions, in the perivascular tissues, and in deeper tissues not contiguous to
host defense. Disseminated intravascular coagulation (DIC) illustrates the link between coagulation and inflammation. In DIC, coagulation pathways are activated, natural inhibitory pathways of coagulation are dysfunctional, and the fibrinolytic system is dysregulated. All of these are direct or indirect consequences of the inflammatory response. The natural inhibitory pathways of coagulation are of particular interest in this intersection of coagulation and inflammation, as potential therapies have been based around these biologic processes (6,7). Coagulation may be initiated in the flowing blood, on the endothelial surface, at endothelial lesions, in the perivascular tissues, and in deeper tissues not contiguous to vascular structures. It may or may not be associated with the formation of fibrin clots (8).
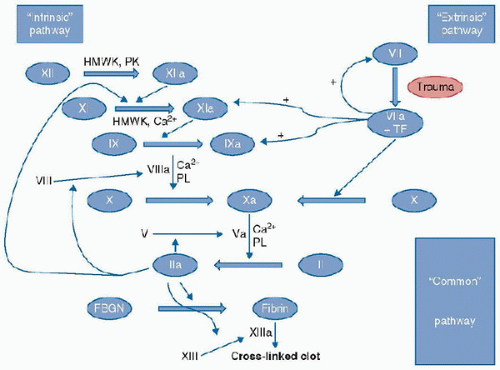 FIGURE 118.2. “Classical” coagulation cascade with crosstalk. Thin lines with “+” indicate enhancement of generation of F.Xia and F.IXa by the F.VIIa/TF complex. HMWK, highmolecular-weight kininogen; PK, prekallekrein; TF, tissue factor; PL, phospholipid; Ca, calcium; Clotting factors: XII, factor XII; XIIa, activated factor XII; XI, factor XI; XI, activated factor XI; IX, factor IX; IXa, activated factor IX; VIIIa, activated factor VIII; VIIa, activated Factor VII; X, factor X; Xa, activated factor Xa; Va, activated factor V; II, prothrombin; IIa, thrombin; FBGN, fibrinogen; XIIIa, activated factor XIII. |
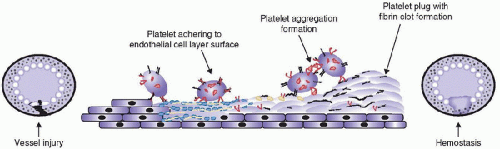 FIGURE 118.3. The role of platelets in mediating primary hemostasis at sites of vascular injury. Platelets are initially activated and express specific adhesion receptors on their surface, followed by adhesion to activated endothelial cells and exposed subendothelial components (e.g., collagen, von Willebrand factor). Subsequent platelet aggregation occurs with the development of a primary platelet plug. Coagulation occurs on the developing platelet plug with the creation of a fibrin clot. |
(APC) also possesses intrinsic immunomodulating properties (see Fig. 118.5). In vitro, APC inhibits tumor necrosis factor-α elaboration from monocytes, blocks leukocyte adhesion to selectins, and influences apoptosis (9). The PC pathway is initiated by the binding of thrombin to thrombomodulin and forms a complex on the surface of endothelial cells. The binding of PC to the endothelial cell PC receptor augments PC activation by the thrombin-thrombomodulin complex more than tenfold. PC activation in sepsis and inflammation is downregulated when the exposure to inflammatory mediators and thrombin causes the endothelial cells to shed their PC receptors. The endothelial cell PC receptor (ECPCR) can also translocate from the plasma membrane to the nucleus and redirect gene expression. The translocation of the PC-receptor-APC complex to the nucleus may account for the ability of APC to modulate inflammatory mediator responses in the endothelium (9).
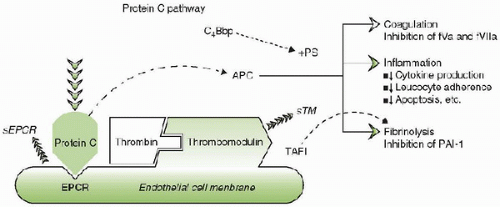 FIGURE 118.4. The interaction of the protein C system with the endothelium. Thrombin bound to thrombomodulin (TM) modifies protein C bound to the endothelial protein C receptor on the cell surface to generate activated protein C (APC). APC acts as a natural anticoagulant by inactivating activated factors V (fVa) and VIII (fVIIIa), modulating inflammation by downregulating the synthesis of proinflammatory cytokines, leukocyte adherence, and apoptosis and enhancing fibrinolysis by inhibiting thrombin-activatable fibrinolysis inhibitor (TAFI) and plasminogen activator inhibitor type-1 (PAI-1). C4Bbp, C4b binding protein; +PS, in the presence of protein S; sTM, soluble thrombomodulin; sEPCR, soluble endothelial cell protein C receptor. |
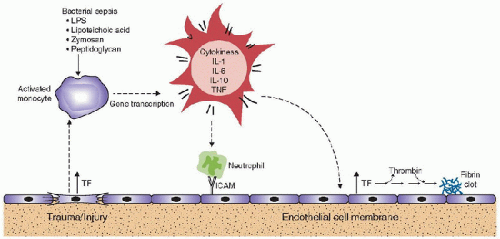 FIGURE 118.5. Inflammation effect on coagulation. Inflammation enhances coagulation through the induction of proinflammatory cytokines that induce tissue factor formation, which in turn decreases activated protein C (APC) formation, leading to enhanced thrombin and fibrin generation. In addition, the decrease in APC allows for greater inhibition of fibrinolysis through the action of plasminogen activator inhibitor type-1 (PAI-1). |
associated with increased levels of various cytokines and acute-phase proteins, abnormal coagulation parameters, increased severity of disease, and poorer outcomes. The regulation of the production of PAI-1 is multifactorial (Fig. 118.6). The 4G/5G insertion/deletion promoter polymorphism of the PAI-1 gene has been shown to affect PAI-1 plasma levels; individuals with the 4G/4G genotype display the highest PAI-1 levels while those with the 5G/5G genotype the lowest (the 4G/5G genotype results in intermediate levels). Differences in PAI-1 levels affect the risk of developing severe complications and death from sepsis. High PAI-1 levels are associated with increased mortality in animal models of sepsis and with increased severity of illness and organ dysfunction scores in septic patients (including children with meningococcal sepsis) (10,11,12). However, the regulation of PAI-1 levels involves more than just the promoters of its synthesis. Activated Protein C stimulates fibrinolysis by forming a tight 1:1 complex with PAI-1 leading to inactivation of PAI-1. High levels of thrombin lead to increased levels of APC, which can complex to PAI-1. This complex is subsequently cleared from the circulation, resulting in PC depletion (11). The increased formation and clearance of these complexes results in PC depletion (11), with the net result being an increased risk for microvascular thrombus formation.
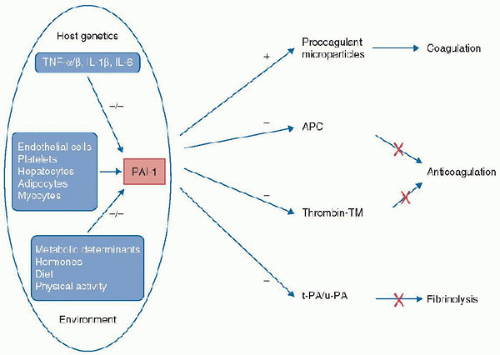 FIGURE 118.6. Plasminogen activator inhibitor type 1 gene polymorphism and sepsis. Genetic and environmental influences on the expression of plasminogen activator inhibitor type 1 (PAI-1) and the importance of PAI-1 in the coagulation and fibrinolysis pathways. TNF-α, tumor necrosis factor-α; APC, activated protein C; TM, thrombomodulin; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator. (Redrawn from Hermans PW, Hazelzet JA. Plasminogen activator inhibitor type 1 gene polymorphism and sepsis. Clin Infect Dis 2005;41(suppl 7):S453-8.) |
abnormalities and are discussed below. Additionally, in specific circumstances, herbal medications may contribute to impaired clot formation or abnormal bleeding (e.g., garlic, ginko, senna, cascara). In trauma and surgical situations, it is important to determine the severity of injury relative to the magnitude of bleeding that follows. A prior history of significant thrombosis (e.g., deep venous thrombosis, pulmonary embolus, stroke) suggests the possibility of a hypercoagulable condition. As thrombotic events are uncommon in children, the occurrence of thrombotic events, such as myocardial infarction in young, adult relatives, should cause the clinician to consider a congenital thrombophilic abnormality. These abnormalities include AT III deficiency, PC or protein S deficiency, factor V Leiden R506Q mutation, the prothrombin G20210A polymorphism/mutation, and (possibly) the C677T mutation/polymorphism of the methylenetetrahydrofolate reductase (MTHFR) gene (discussed below). In addition, vasculitis associated with an autoimmune disorder such as systemic lupus erythematosus (SLE) must be considered in the evaluation of a child with an unexplained clot. In all cases, the family history is important in attempting to separate congenital from acquired disorders.
 causes of generalized bleeding in critically ill children include sepsis-related DIC, massive transfusion syndrome (discussed below), severe liver dysfunction, undiagnosed hemophilia, battered child syndrome, and vitamin K deficiency in newborns or older children with malabsorption (18,19,20). In young infants (<3 months of age), the coagulation system is often not yet mature, and prolongation of the prothrombin time (PT) or activated partial thromboplastin time (aPTT) may not reflect an abnormality in hemostasis (21). Consultation with a pediatric hematologist may be indicated when trying to interpret “abnormal” results in this age group.
causes of generalized bleeding in critically ill children include sepsis-related DIC, massive transfusion syndrome (discussed below), severe liver dysfunction, undiagnosed hemophilia, battered child syndrome, and vitamin K deficiency in newborns or older children with malabsorption (18,19,20). In young infants (<3 months of age), the coagulation system is often not yet mature, and prolongation of the prothrombin time (PT) or activated partial thromboplastin time (aPTT) may not reflect an abnormality in hemostasis (21). Consultation with a pediatric hematologist may be indicated when trying to interpret “abnormal” results in this age group. disorder should address several basic questions. Is the process localized or diffuse? Is it related to an anatomic or surgical lesion (e.g., bleeding localized to an area of trauma or recent surgical site not associated with generalized bleeding suggests a discrete anatomic cause for the bleeding rather than the presence of a generalized hemorrhagic diathesis)? Is the bleeding primarily muco-cutaneous? Finally, when appropriate, are there signs of thrombosis (either arterial or venous)? The answers to these questions may provide clues to the cause of the problem.
disorder should address several basic questions. Is the process localized or diffuse? Is it related to an anatomic or surgical lesion (e.g., bleeding localized to an area of trauma or recent surgical site not associated with generalized bleeding suggests a discrete anatomic cause for the bleeding rather than the presence of a generalized hemorrhagic diathesis)? Is the bleeding primarily muco-cutaneous? Finally, when appropriate, are there signs of thrombosis (either arterial or venous)? The answers to these questions may provide clues to the cause of the problem.In most instances, measurement of the platelet count, fibrinogen level, PT, aPTT, and TT should be sufficient for determining the correct diagnosis. An unnecessary use of laboratory resources may be avoided by using these five screening tests and only ordering further, more specific, testing when a definitive diagnosis is necessary. Several major categories of hemorrhagic disorders and the tests that are characteristically abnormal in each are summarized in Table 118.2.
TABLE 118.2 COAGULATION DISORDERS AND ASSOCIATED LABORATORY FINDINGS | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
 being present. While hyperfibrinogenemia (>400 mg/dL) and persistent elevations of F.VIII (>400%) have been associated with an increased risk of thrombosis in adults, both may be elevated by acute inflammation. Consequently, the finding of elevations of these clotting factors in a patient who has experienced an unexpected thrombotic event may not necessarily indicate that the cause of the event was an elevation in either factor. Without prior samples, it is impossible to determine if the elevation is the consequence of the thrombosis or was present prior to its development and potentially causative.
being present. While hyperfibrinogenemia (>400 mg/dL) and persistent elevations of F.VIII (>400%) have been associated with an increased risk of thrombosis in adults, both may be elevated by acute inflammation. Consequently, the finding of elevations of these clotting factors in a patient who has experienced an unexpected thrombotic event may not necessarily indicate that the cause of the event was an elevation in either factor. Without prior samples, it is impossible to determine if the elevation is the consequence of the thrombosis or was present prior to its development and potentially causative.when consumption exceeds production (29). The conditions associated with DIC are generally the same in adults and children and include the wide variety of disorders with the ability to initiate coagulation (Table 118.3). The mechanisms involved in these conditions either activate procoagulant proteins enzymatically or cause the release of TF, which then triggers coagulation.
TABLE 118.3 CONDITIONS ASSOCIATED WITH DISSEMINATED INTRAVASCULAR COAGULATION | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
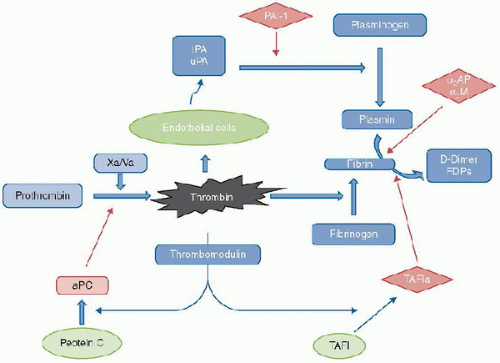 FIGURE 118.7. Fibrinolysis. Thrombin, generated from prothrombin by the action of the Xa/Va prothrombinase complex activates endothelial cells to produce plasminogen activators. These, in turn, cleave plasminogen to form plasmin, which degrades fibrin (formed by the action of thrombin on fibrinogen) to D-dimer fragments and other fibrinogen degradation products. Inhibitors of fibrinolysis (in diamonds) include PAI-1, which inhibit the actions of tPA and uPA, and α2AP, α2M, and TAFI, which inhibit plasmin. Thrombin, when bound to thrombomodulin on the surface of endothelial cells, activates TAFI to its active form and produces activated Protein C an important vitamin K-dependent protein with anticoagulant and anti-inflammatory properties. α2AP, alpha-2-antiplasmin (alpha-2-plasmin inhibitor); α2M, alpha-2-macroglobulin; tPA, tissue-type plasminogen activator; uPA, urine-type plasminogen activator; PAI-1, plasminogen activator inhibitor type-1; FDPs, fibrin degradation products; Xa, activated factor X; Va, activated factor V; aPC, activated Protein C; TAFI, thrombin-activatable fibrinolysis inhibitor; TAFIa, activated TAFI. |
Whether the presentation of DIC is thrombotic, hemorrhagic, or “compensated” (that is, laboratory results consistent with DIC without bleeding), microthrombosis likely contributes to the development and progression of multiorgan failure.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


