Channelopathies and Their Possible Relation to Migraines
Dennis E. Bulman
Daniela Pietrobon
Ion channels are part of a large family of macromolecules whose functions include the control and maintenance of electrical potential across cell membranes, generation of electrical signals, secretion, and signal transduction (25). The superfamily of voltage-gated ion channels includes the K+, Na+, and Ca2+. These cation channels are related, sharing a fundamental design consisting of a set of six potentially membrane-spanning segments (S1 to S6). The six segments comprise a domain present only once in the K+ channels, but is repeated four times within the α1 subunit of the Na+ and Ca2+ channels. S4 is thought to function as the voltage sensor and contains basic residues at every third or fourth position (25,27). The amino acid sequence of the individual voltage-gated ion channels are highly conserved across species, with some regions of the channel being conserved between humans and Drosophila. Generally, a high degree of sequence conservation across species has been considered evidence in favor of strong selective pressure on these channels. Mutations in these voltage-gated ion channel genes have been shown to cause or have been implicated in a number of episodic disorders, including periodic paralysis, episodic ataxia, long QT syndrome, paroxysmal dyskinesia, and familial hemiplegic migraine (FHM). Correlation of the various mutations with the clinical phenotype is providing insight into the pathophysiology of these channel proteins. Interestingly, different mutations within the same ion channel gene may cause quite distinct clinical disorders; conversely, mutations in different ion channel genes may result in very similar phenotypes (genetic heterogeneity). The role played by ion channels in the pathogenesis of neuromuscular and neurologic disease represents a paradigm for the role of ion channels in episodic disorders such as migraine.
FHM is a rare, autosomal-dominant form of migraine with aura. The Headache Classification Committee of the International Headache Society states that the diagnosis of FHM is based on the presentation of hemiparesis during the aura phase, preceding the headache phase, and identical attacks must occur in at least one first- or second-degree family member (3). Almost invariably, at least three aura symptoms (and often four) are present (usually in the temporal order: visual, sensory, motor, and aphasic). In general, the duration of headache is longer in FHM patients than in migraine with aura patients (50). In contrast to other types of migraine, some FHM patients can have atypical severe attacks with impairment of consciousness (coma), prolonged hemiplegia lasting several days, or both. Some patients have permanent signs of disease, such as nystagmus and ataxia between attacks. Mutations in the genes CACNA1A at 19p13 or ATP1A2 at 1q23 are responsible for FHM1 (OMIM 141500) and FHM2 (OMIM 602481), respectively. At least one other locus for FHM must exist because families not linked to either the FHM1 or FHM2 loci have been described (15). With the identification of two genes, responsible for FHM, there is a move toward the incorporation of molecular genetic analysis in its classification (3,21). Clinically, there are few differences in symptoms between FHM owing to mutations in the CACNA1A gene and the other cases of FHM (48).
Mutations in the CACNA1A gene are also responsible for episodic ataxia type 2 (EA-2) and spinocerebellar ataxia type 6 (SCA6) (42,61). Individuals with familial EA-2 experience discrete episodes of pancerebellar disturbances with dysarthria, titubations, dysmetric limb movements, severe truncal and gait ataxia; headaches, vertigo, and nausea were reported in a subset of patients (17,20,60). Patients with EA-2 may have an associated migraine (nonhemiplegic) that presents after the onset of the ataxic symptoms. Interictal examination reveals often persistent nystagmus and residual mild cerebellar incoordination. Atrophy of the cerebellar vermis has been described in some families with EA-2 (56), but not in others (6,13,38,59). In a few cases epilepsy has been reported in addition to EA-2 (11,30). Shortly after the identification
of the FHM/EA-2 gene defects, the trinucleotide repeat expansion disorder SCA6 was found to be caused by a CAG expansion within CACNA1A (61). SCA6 is a mild but slowly progressive cerebellar ataxia of the limbs and gait. Although mutations in CACNA1A may also cause EA-2, SCA6, or epilepsy, FHM1 patients may exhibit symptoms that overlap with these other disorders. Overall, this may not be surprising given that the disorders are caused by mutations in the same gene.
of the FHM/EA-2 gene defects, the trinucleotide repeat expansion disorder SCA6 was found to be caused by a CAG expansion within CACNA1A (61). SCA6 is a mild but slowly progressive cerebellar ataxia of the limbs and gait. Although mutations in CACNA1A may also cause EA-2, SCA6, or epilepsy, FHM1 patients may exhibit symptoms that overlap with these other disorders. Overall, this may not be surprising given that the disorders are caused by mutations in the same gene.
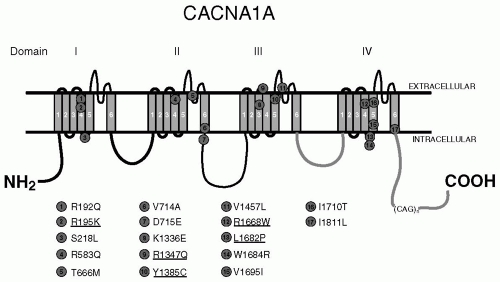 FIGURE 32-1. Predicted structure of the P/Q-type calcium channel α1A-subunit illustrating the four homology domains each composed of six transmembrane segments (S1 to S6). Mutations within CACNA1A responsible for FHM are indicated and those mutations for which functional analysis has not been performed are underlined. |
FAMILIAL HEMIPLEGIC MIGRAINE TYPE 1
In 1996, mutations responsible for FHM were identified in the CACNA1A gene (42). The CACNA1A gene encodes the pore-forming α1-subunit of the neuronal voltage-gated Cav2.1 (P/Q-type) calcium channel and maps to chromosome 19p13. CaV2.1 channels are located in presynaptic terminals and somatodendritic membranes throughout the brain, and play a prominent role in controlling neurotransmitter release (10,58). The somatodendritic localization points to additional postsynaptic roles, for example, in neural excitability and gene expression. CaV2.1 channels are expressed in all brain structures that have been implicated in the pathogenesis of migraine, including the cerebral cortex, the trigeminal ganglia, and brainstem nuclei involved in the central control of nociception (for a review see Pietrobon and Striessnig [43]). Their expression is particularly high in the cerebellum.
Roughly half of the cases of FHM are caused by mutations in CACNA1A (29), and it appears that cerebellar ataxia may occur in 50% of the cases of FHM1. In addition, some cases of FHM1 have been shown to be triggered by mild head trauma and also be associated with coma (14,34,49). To date, there are 17 known mutations within CACNA1A that result in FHM1, all of which are caused by amino acid substitutions of conserved amino acids in important functional regions of the channel, including the pore lining and the voltage sensors (Fig. 32-1). Among the most recurring mutations, the T666M mutation appears to demonstrate the highest penetrance (98%), severe attacks with coma (50%) and nystagmus (86%) (14), and the R583Q mutation appears to have the highest penetrance of gait ataxia (81%) in the absence of any nystagmus (14).
Functional Consequences of FHM1 Mutations
The functional consequences of 12 FHM1 mutations have been investigated in heterologous expression systems (oocytes or mammalian cell lines) expressing recombinant CaV2.1 channels (23,36,37,41,51). Because CaV2.1 channel expression is almost exclusively restricted to neuronal cells, five of these FHM1 mutations have also been investigated in neurons from CaV2.1α1−/− mice
expressing human CaV2.1α1 subunits (8,51). Recently, the generation of a knockin mouse carrying the R192Q FHM1 mutation allowed the first analysis of mutant channels expressed at their endogenous level in neurons (54). The studies in heterologous expression systems showed that the FHM1 mutations alter many biophysical properties of human CaV2.1 channels, in a complex way. In some cases (e.g., in the case of the channel-inactivation properties) the effects are different (e.g., increased, decreased, or unaffected inactivation during train of pulses) depending on the mutation. However, an important consistent effect of FHM1 mutations was revealed by studying their effect on the function of single human CaV2.1 channels, using single channel patch-clamp recordings (23,51,52) (and our unpublished observations). All eight FHM1 mutations analyzed so far at the single-channel level produced an enhanced Ca2+ influx through human CaV2.1 channels over a broad voltage range, reflecting an increased channel open probability, mainly owing to a shift to lower voltages of channel activation. As a consequence of this shift, mutant human CaV2.1 channels open at lower voltages and more readily than wild-type (wt) channels, and Ca2+ influx through mutant channels can occur in response to small depolarizations insufficient to open wt channels. The analysis of the voltage-dependence of the whole-cell current, produced by the 12 FHM1 mutants expressed in heterologous systems and the 5 mutants expressed in CaV2.1α1−/− neurons, essentially confirm and support the single-channel phenotype of mutant human CaV2.1 channels, because it revealed a consistent (and in the majority of cases significant) shift to lower voltages of current activation. Also the whole-cell CaV2.1 current in neurons of the R192Q knockin mouse was found to activate at lower voltages than in wt neurons (54). Moreover, in agreement with the gain-of-function phenotype of mutant human CaV2.1 channels, the whole-cell CaV2.1 current density in the R192Q knockin neurons was larger than in wt neurons in a broad voltage range (for voltages lower than −10 mV), and similar to that of wt at voltages higher than −10 mV. The number of functional channels per unit neuronal membrane area was similar in knockin and wt mice, in contrast with the alterations found in transfected cells where there were decreased numbers of mutant R192Q channels in neurons and increased number in HEK293 cells (23,51). In transfected cells overexpressing CaV2.1 channels, all the FHM1 mutations analyzed produced alterations of the number of functional channels in the membrane, leading to a consistent decrease of the whole-cell CaV2.1 current density at high voltages in transfected neurons (8,51) and to opposite effects on the whole-cell CaV2.1 current density depending on the mutation in transfected HEK293 cells (23). The confusing decreased whole-cell CaV2.1 current density measured at high voltages in transfected neurons was not observed in the knockin neurons that do not overexpress the mutant channels. Therefore, the alterations in the number of functional channels produced by the mutations in transfected cells may be an artifact caused by overexpression.
expressing human CaV2.1α1 subunits (8,51). Recently, the generation of a knockin mouse carrying the R192Q FHM1 mutation allowed the first analysis of mutant channels expressed at their endogenous level in neurons (54). The studies in heterologous expression systems showed that the FHM1 mutations alter many biophysical properties of human CaV2.1 channels, in a complex way. In some cases (e.g., in the case of the channel-inactivation properties) the effects are different (e.g., increased, decreased, or unaffected inactivation during train of pulses) depending on the mutation. However, an important consistent effect of FHM1 mutations was revealed by studying their effect on the function of single human CaV2.1 channels, using single channel patch-clamp recordings (23,51,52) (and our unpublished observations). All eight FHM1 mutations analyzed so far at the single-channel level produced an enhanced Ca2+ influx through human CaV2.1 channels over a broad voltage range, reflecting an increased channel open probability, mainly owing to a shift to lower voltages of channel activation. As a consequence of this shift, mutant human CaV2.1 channels open at lower voltages and more readily than wild-type (wt) channels, and Ca2+ influx through mutant channels can occur in response to small depolarizations insufficient to open wt channels. The analysis of the voltage-dependence of the whole-cell current, produced by the 12 FHM1 mutants expressed in heterologous systems and the 5 mutants expressed in CaV2.1α1−/− neurons, essentially confirm and support the single-channel phenotype of mutant human CaV2.1 channels, because it revealed a consistent (and in the majority of cases significant) shift to lower voltages of current activation. Also the whole-cell CaV2.1 current in neurons of the R192Q knockin mouse was found to activate at lower voltages than in wt neurons (54). Moreover, in agreement with the gain-of-function phenotype of mutant human CaV2.1 channels, the whole-cell CaV2.1 current density in the R192Q knockin neurons was larger than in wt neurons in a broad voltage range (for voltages lower than −10 mV), and similar to that of wt at voltages higher than −10 mV. The number of functional channels per unit neuronal membrane area was similar in knockin and wt mice, in contrast with the alterations found in transfected cells where there were decreased numbers of mutant R192Q channels in neurons and increased number in HEK293 cells (23,51). In transfected cells overexpressing CaV2.1 channels, all the FHM1 mutations analyzed produced alterations of the number of functional channels in the membrane, leading to a consistent decrease of the whole-cell CaV2.1 current density at high voltages in transfected neurons (8,51) and to opposite effects on the whole-cell CaV2.1 current density depending on the mutation in transfected HEK293 cells (23). The confusing decreased whole-cell CaV2.1 current density measured at high voltages in transfected neurons was not observed in the knockin neurons that do not overexpress the mutant channels. Therefore, the alterations in the number of functional channels produced by the mutations in transfected cells may be an artifact caused by overexpression.
Two important conclusions can be drawn from the studies of the functional consequences of FHM1 mutations described. First, the FHM1 mutations lead to gain-of-function of human neuronal CaV2.1 channels. Second, as a general principle in the study of channelopathies, relying on work from transfected cells overexpressing the channels of interest is not good enough if one wishes to understand the disease mechanisms. Transfected cells can be used reliably to study the effect of the mutations on the single-channel function, but not on cellular functions such as neuronal excitability or synaptic transmission that depend on the number of functional channels in the membrane. Animal models appear necessary to answer the challenging question of how the changes in channel function produced by the mutations lead to the specific cellular dysfunctions and to the symptoms of the disease.
To begin to answer the question of how gain-of-function of CaV2.1 channels lead to FHM1, van den Maagdenberg et al. (54) investigated in the R192Q knockin mouse (i) synaptic transmission at the neuromuscular junction (NMJ), a synapse where neurotransmitter release depends predominantly on CaV2.1 channels, as occurs in most cortical excitatory synapses releasing glutamate, and (ii) the in vivo threshold for initiation and rate of propagation of cortical spreading depression (CSD), the likely generator of migraine aura (7,22). They found increased neurotransmission at the NMJ in conditions of low release probability where saturation of the synaptic Ca2+ sensors was not reached (evoked release at low Ca2+ and spontaneous release). Because many brain excitatory synapses are characterized by a low probability of release in response to an action potential (22,44), the knockin NMJ data suggest enhanced glutamate release at cortical excitatory synapses consequent to gain-of-function of CaV2.1 channels containing FHM1 mutations.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


