KEY POINTS
Correct diagnoses and understanding of arrhythmia mechanisms are crucial to successful arrhythmia management.
The hemodynamic effects of an arrhythmia are important in developing an appropriate treatment strategy.
Predisposing conditions and reversible causes should be recognized and corrected.
Knowledge of antiarrhythmics for prophylaxis, acute management, and long-term management are necessary for successful arrhythmia management.
The proarrhythmic potential of antiarrhythmic drugs must be recognized and preventative measures should be taken whenever possible.
Knowledge of antiarrhythmic drug pharmacokinetics and pharmacodynamics and the impact of multisystem organ disease on these parameters are important in preventing drug toxicity.
The intensivist should be skilled in the implantation of temporary pacing systems, external cardioversion, and defibrillation.
Role of cardiac pacing, electrical cardioversion, and defibrillation should be clear to physicians managing patients with arrhythmias.
Recognition of malfunctioning temporary and permanent implantable pacemakers or cardioverter defibrillators is crucial in the management of patients with such devices.
Cardiac arrhythmias are common in the critical care setting. Many arrhythmias detected are benign, may occur in healthy individuals and require no investigation or treatment, for example, sinus tachycardia, sinus bradycardia, Mobitz type I second-degree AV block or premature atrial and ventricular beats. At times, the arrhythmia may be the clue to a sick patient, such as sinus tachycardia in a patient developing sepsis or atrial fibrillation due to a pulmonary embolus. Correct diagnosis and understanding arrhythmia mechanisms as well as knowledge of antiarrhythmic drug pharmacology and nonpharmacologic therapies of arrhythmias are crucial for successful arrhythmia management. This chapter focuses on the mechanisms, investigation, and management of the most common, clinically significant arrhythmias encountered.
TACHYARRHYTHMIAS
Tachycardia mechanisms have been classified as due to abnormalities of impulse formation or impulse conduction.1-3 Abnormalities of impulse formation may be due to normal automaticity, abnormal automaticity or triggered activity occurring within atrial or ventricular muscle tissue or the specialized conduction system (Fig. 36-1A).1,3 Natural pacemaker cells are found in the sinus node, parts of the atria, the atrioventricular node, and the His-Purkinje system. These cells exhibit phasic spontaneous depolarization during diastole, resulting in an action potential when the threshold potential is reached.1 Although in the normal heart, the sinus node is the dominant pacemaker, subsidiary pacemakers may become dominant under certain conditions, for example, sympathetic stimulation or digitalis toxicity (Fig. 36-1). Normal atrial and ventricular muscle maintains a high negative resting potential (−90 mV) and only depolarizes when stimulated. Under certain pathophysiologic conditions, for example, electrolyte abnormalities or ischemia, the resting membrane potential may decrease (−60 mV) and cells may now spontaneously depolarize.
FIGURE 36-1
Mechanisms of cardiac arrhythmias—enhanced or abnormal automaticity, triggered activity or reentry. A. The rate of phase 4 depolarization may increase causing the myocardial cell to reach the threshold potential (TP) earlier and spontaneously depolarize. In diseased tissue, the resting potential (RP) may be elevated and the time to reach TP may then be shortened (not shown). B. Early afterdepolarizations (EADs) develop late on phase 3 of repolarization of the action potential and if they reach threshold may trigger a depolarization. This is the mechanism of torsade de pointes ventricular tachycardia. C. Delayed afterdepolarizations (DADs) occur in diastole after repolarization is complete. If they reach threshold, they may trigger a depolarization. This is the mechanism associated with digitalis toxicity. D. Reentry is associated with an area of anatomic or functional unidirection block (speckled area) and a region of slow conduction (represented by zigzag arrow). A propagated impulse blocks in one area of tissue, passes through an area of slow conduction and then conducts retrogradely through the original area of conduction block, which has now repolarized.
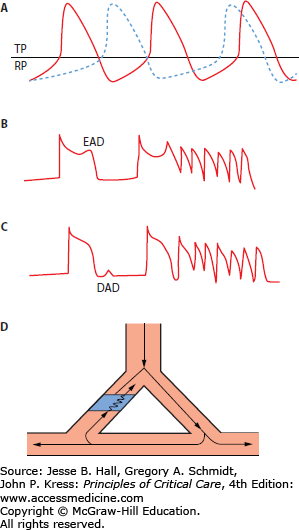
Triggered activity is caused by afterdepolarizations that occur early in repolarization (early afterdepolarizations [EADs]) or after repolarization is complete (delayed afterdepolarizations [DADs]) (Fig. 36-1B).1,3 EADs may reach threshold to activate the slow inward current generating a new action potential and this cycle may repeat generating a sustained tachycardia. EADs are believed to be the mechanism of torsade de pointes ventricular tachycardia (VT).3 DADs initiate a triggered response only when their amplitude reaches a critical threshold (Fig. 36-1C). Increasing the heart rate or the prematurity of an extrastimulus increases the amplitude of a DAD thus increasing the probability of inducing a tachycardia. DADs are thought to cause arrhythmias secondary to digitalis toxicity and in the setting of heart failure.
Abnormalities of impulse conduction are primarily due to reentry (Fig. 36-1).2 Reentry requires an area of fixed or functional unidirectional block in one pathway, slow conduction in an alternate pathway and return of the impulse along the original path after this tissue has recovered excitability (Fig. 36-1D). Reentry is the mechanism of many types of supraventricular tachycardias (atrioventricular node reentry or in the Wolff-Parkinson-White syndrome) and scar-related ventricular tachycardia (VT) that occurs following a myocardial infarction. Table 36-1 summarizes the mechanisms of common tachyarrhythmias.
Mechanisms of Common Tachycardias
| Arrhythmia | Mechanism(s) |
|---|---|
| Supraventricular Tachycardia | |
| AV node | Reentry |
| Accessory pathway | Reentry |
| Atrial tachycardia | Enhanced automaticity or reentry |
| Atrial flutter | Reentry |
| Atrial fibrillation | Onset—triggered activity |
| Maintenance—multiple wavelet reentry | |
| Ventricular Tachycardia | |
| Monomorphic post MI | Reentry |
| Catecholamine sensitive | Triggered activity |
| Torsade de pointes | Triggered activity |
| Ventricular fibrillation | Multiple wavelet reentry |
ANTIARRHYTHMIC DRUG THERAPY
Antiarrhythmic drug therapy may be required for the termination of tachyarrhythmias and prevention of recurrence. Knowledge of the potential mechanisms of an arrhythmia as well as the pharmacology of antiarrhythmic drugs is important in selecting appropriate drug therapy. Since the critical care patient often has multisystem disease, special attention to factors that influence drug absorption, protein binding, drug metabolism and excretion as well as knowledge of potential drug interactions is essential.4
The cardiac action potential varies strikingly in duration and morphology in specific regions of the heart reflecting differences in ion channel expression or differences in modulators of ion channel function.4 Antiarrhythmic drugs may therefore exert different effects on these various cardiac cells. Moreover, the effects of antiarrhythmic drugs may be modified in diseased cardiac tissue. For example, the effects of sodium channel blockers may be exaggerated in ischemic myocardium and the effects of potassium channel blockers may be exaggerated in the setting of left ventricular hypertrophy.4 Antiarrhythmic drugs are classified most commonly by their dominant mechanism of action.4-6 However, this drug classification scheme is imperfect as many of these drugs have effects on multiple ion channels and/or cell membrane receptors (Table 36-2). Efficacy of one drug does not predict the efficacy of another drug in the same class.
Pharmacodynamics of Antiarrhythmic Drugs
| Recovery From Sodium Channel Block | K+ Channels | Receptors | |
|---|---|---|---|
| Class I Sodium Channel Blockers | |||
| Class IA | |||
| Disopyramide | Intermediate | ↓Ito, ↓IKr | Inhibits muscarinic receptors |
| ↓IK(ATP) | |||
| Quinidine | Intermediate | ↓Ito, ↓IKr | Inhibits alpha and muscarinic receptors |
| Procainamide | Intermediate | ||
| N-acetyl procainamide | – | ↓IKr | |
| Class IB | |||
| Lidocaine | Rapid | – | – |
| Mexiletine | Rapid | – | – |
| Class IC | |||
| Flecainide | Slow | ↓IKr, ↓IKur | |
| Propafenone | Slow | ↓IKr, ↓IKur | Inhibits β-receptors |
| Class II β-Adrenergic Receptor Blockers | |||
| Atenolol | – | β1-Receptor blocker | |
| Bisoprolol | – | β1-Receptor blocker | |
| Carvedilol | – | β1-Receptor blocker | |
| α-Receptor blocker | |||
| Metoprolol | – | β1-Receptor blocker | |
| Nadolol | – | Nonselective β-blocker | |
| Propranolol | Rapid | Nonselective β-blocker | |
| Class III Drugs That Prolong Repolarization | |||
| Amiodarone | Rapid | ↓IKr | Inhibits α- and β-receptors |
| Calcium channel blocker | |||
| Dofetilide | – | ↓IKr | |
| Sotalol | – | ↓IKr | Nonselective β-blocker |
| Dronedarone | Rapid | ↓IKr | Inhibits α- and β-receptors |
| Calcium channel blocker | |||
| Class IV Calcium Channel Blockers | |||
| Diltiazem | – | – | – |
| Verapamil | Rapid | – | – |
| Digoxin | – | – | Blocks-Na+-K+ ATPase |
Class I drugs block the inward sodium current resulting in slowing of conduction in atrial and ventricular muscle.6 Their subclassification is based on the rate of recovery from sodium channel block. Class IA antiarrhythmic drugs have an intermediate rate of recovery of sodium channel block and may cause prolongation of the QRS duration at physiologic heart rates. Class IB drugs have a rapid recovery from sodium channel block and thus slow ventricular conduction only at very rapid heart rates. Class IA and IB drugs are used infrequently. Class IC drugs have a slow rate of recovery from sodium channel block and may cause significant prolongation of the QRS duration at resting heart rates. Class IA and IC drugs also block a variety of potassium channels that causes prolongation of the atrial and ventricular action potential durations. This may manifest as prolongation of the QT interval. Class II drugs are β-adrenergic receptor blocking drugs.7 Some of these agents are selective β1-receptor blockers (Table 36-2). Class III drugs are predominantly potassium channel blockers and prolong repolarization in atrial and ventricular muscle.8,9 They can cause significant prolongation of the QT interval. Class IV drugs are calcium channel blocking drugs that reduce intracellular calcium concentrations.10
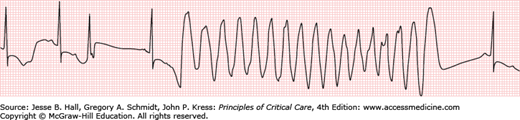
The mechanism(s) of action of commonly used antiarrhythmic drugs are listed in Table 36-2. Many of these agents are used to treat both supraventricular and ventricular arrhythmias. Perhaps the most serious adverse response to antiarrhythmic drugs is the risk of ventricular proarrhythmia.7,11 Proarrhythmia is defined as provocation of a new arrhythmia or worsening of an existing arrhythmia during therapy with a drug at concentrations not considered to be toxic. Class IA and IC drugs may cause excessive slowing of conduction in diseased atrial or ventricular muscle tissue that may exacerbate the clinical arrhythmia, for example, atrial flutter or VT. These drugs increase the risk of sudden cardiac death in patients following myocardial infarction. Thus, Class I antiarrhythmic drugs are contraindicated in patients with ischemic heart disease and a prior myocardial infarction because of the risk of ventricular proarrhythmia. Drugs that cause excessive prolongation of the QT interval may cause torsade de pointes VT (Fig. 36-2).4,7,11 Torsade de pointes VT may occur in 1% to 8% of patients exposed to QT interval prolonging antiarrhythmic drugs.4
The pharmacokinetic characteristics of commonly used antiarrhythmic drugs are summarized in Table 36-3.6,12-15 Drug dosing, adverse effects and potential interactions are listed in Table 36-4.4,6,8,10,12,16,17 Adverse effects may develop from pharmacokinetic or pharmacodynamic drug interactions. Pharmacokinetic drug interactions develop when one drug modifies the absorption, distribution, metabolism, or elimination of a second drug, for example, warfarin and amiodarone or digitalis and quinidine.4,12 Pharmacodynamic interactions occur when a drug or condition increases or reduces the pharmacologic effect of a drug without changing plasma drug concentrations, for example, increased protein binding of propafenone, verapamil, or lidocaine secondary to elevated α1-acid glycoprotein following myocardial infarction.12 Several enzymes in the cytochrome P450 family are responsible for drug metabolism. Some drugs may inhibit or induce these enzymes resulting in important drug interactions (Table 36-4).4,12,13 Mutations or polymorphisms of genes encoding enzymes responsible for drug metabolism may cause important drug interactions.18,19
Pharmacokinetic Characteristics of Antiarrhythmic Drugs
| Drug | Bioavailability (%) | Binding to Plasma Proteins (%) | Renal Elimination of Unchanged Drug (%) | Plasma Half Life (h) | Active Metabolites | Hepatic Metabolism |
|---|---|---|---|---|---|---|
| Class IA | ||||||
| Disopyramide | 83 ± 11 | 28%-68%a | 55 ± 6 | 6.0 ± 1.0 | Racemic mixture | |
| Quinidine | 70-80 | 87 ± 3 | 18 ± 5 | 6.2 ± 1.8 | 3-Hydroxy quinidine | CYP3A4 |
| Procainamide | 83 ± 16 | 16 ± 5 | 67 ± 8 | 3.0 ± 0.6 | NAPA | NAT2 acetylationb |
| Class IB | ||||||
| Lidocaine | Parenteral administration only | 70 ± 5 | 2 ± 1 | 1.8 ± 0.4 | Monoethylglycylxylidide | CYP3A4 |
| Mexiletine | 87 ± 13 | 63 ± 3 | 4-15 | 9.2 ± 2.1 | – | CYP1A2 |
| CYP2D6 | ||||||
| Class IC | ||||||
| Flecainide | 70 ± 11 | 61 ± 10 | 43 ± 3 | 11 ± 3 | – | CYP2D6 |
| Propafenone | 5-50a | 85-95 | <1 | 5.5 ± 2.1 | 5-Hydroxy-propafenone | CYP2D6 |
| Class II | ||||||
| Atenolol | 50-60 | <5 | 85-100 | 5-7 | Racemic mixture | – |
| Bisoprolol | 90 | 30 | 50 | 11-17 | Racemic mixture | CYP2D6 |
| Carvedilol | 25 | 95 | <2 | 2.2 ± 0.3 | Racemic mixture | – |
| Metoprolol | 38 ± 1.4 | 11 ± 1 | 10 ± 3 | 3.2 ± 0.2 | Racemic mixture | CYP2D6 |
| Propranolol | 26 ± 10 | 87 ± 6 | <0.5 | 3.9 ± 0.4 | Racemic mixture, hydroxyl propranolol | CYP2D6 |
| CYP1A2 | ||||||
| Class III | ||||||
| Amiodarone | 46 ± 22 | 99.9 ± 0.1 | 0 | 25 ± 12 days | Desethylamiodarone | CYP3A4 |
| Dofetilide | 96(83-108) | 64 | 52 ± 2 | 7.5 ± 0.4 | CYP3A4 | |
| Sotalol | 90-100 | None | >90 | 8 ± 3 | Racemic mixture | – |
| Dronedarone | 4-15 | 98 | <6 | 13-19 | N-debutyl metabolite | CYP3A4 |
| Class IV | ||||||
| Diltiazem | 38 ± 11 | 78 ± 3 | <4 | 4.4 ± 1.3 | Desacetyl diltiazem | CYP3A4 |
| N-desmethyl diltiazem | ||||||
| Verapamil | 22 ± 8 | 90 ± 2 | <3 | 4.0 ± 1.5 | Racemic mixture | CYP3A4 |
| CYP2C9 | ||||||
| Digoxin | 7.0 ± 13 | 25 ± 5 | 60 ± 11 | 39 ± 13 | – | – |
Antiarrhythmic Drug Dosing and Adverse Effects
| Drug | Dosage | Dosage Adjustment | Adverse Effects | Drug Interactions |
|---|---|---|---|---|
| Class IA | ||||
| Quinidine gluconate (sustained release) | 250 mg PO q8h | ↓ Initial dose 50% + ↑ dosing interval to q12h in renal failure | Diarrhea, stomach cramps, tinnitus; torsade de pointes VT tinnitus, fever, rash, thrombocytopenia, hemolytic anemia torsade de pointes VT | ↓ Digoxin dose by 50% monitor INR if on warfarin |
| ↑ By 250 mg doses if QTc <460 ms | Active metabolites accumulate in renal failure but therapeutic blood monitoring of them is not readily available | ↓ Dose of β-blocker; amiodarone, cimetidine, diltiazem, propranolol, and verapamil all of which may increase quinidine concentrations | ||
| ↓ Dose if QTc ≥ 500 ms. Discontinue if QTc ≥ 550 ms. Max. dose 1 g PO q8h | Careful monitoring of the ECG intervals should guide dosing decisions | |||
| Procainamide SR | 250 mg PO q6h | Metabolism depends on rate of acetylation. The active metabolite NAPA accumulates in fast acetylators and in renal failure. Monitor procainamide + NAPA levels and keep sum <80 µM; monitor ECG intervals | Agranulocytosis, rash, fever, SLE syndrome, torsade de pointes VT | Amiodarone, cimetidine, propranolol may increase procainamide concentrations |
| ↑ By 250 mg increments if QTc <460 ms | ||||
| Max. dose 1 g PO q6h | ||||
| ↓ Dose if QTc ≥ 500 ms. | ||||
| Discontinue if QTc > 550 ms | ||||
| Procainamide IV | 750-1000 mg loading; 15-17 mg/kg at 20 mg/min; maintenance 1-4 mg/min | Hypotension | ||
| Disopyramide | 100 mg PO q8h | ↓ Initial dose 50% and ↑ dosing interval q12h in renal failure | Urinary retention, blurred vision constipation, dry mouth, worsening heart failure, torsade de pointes VT | |
| Sustained release 150-250 mg q12h | ||||
| ↑ By 100 mg increments if QTc <460 ms | ||||
| Max dose 300 mg PO q8h ↓dose if QTc ≥ 500 ms | ||||
| Discontinue if QTc ≥ 550 ms | ||||
| Class IB | ||||
| Lidocaine IV | 1.5 mg/kg loading; 1-4 mg/min maintenance | ↓ Dose in CHF | Numbness, paresthesia, slurred speech, altered consciousness | Propranolol, metoprolol, cimetidine increase lidocaine concentrations |
| Mexiletine | 150-300 mg PO q8h | Nausea, stomach cramps tremor, blurred vision, ataxia, confusion | Cimetidine, quinidine increase mexiletine concentrations | |
| Class IC | ||||
| Flecainide | 50 mg PO q12h | ↓ Initial dose 50% in renal failure; titrate dose based on QRS intervals | Tremor, blurred vision, headache, ataxia, CHF,VT proarrhythmia | Amiodarone, cimetidine, propranolol, quinidine increase flecainide concentrations |
| ↑ By 25-50 mg increments | ||||
| Max. dose 200 mg PO q12h | ||||
| ↓ Dose if QRS prolonged >20% from baseline | ||||
| Propafenone | 150 mg PO q8-12h | ↓ Initial dose 50% in renal and hepatic failure and ↑ dosing interval to q12h | Constipation, dizziness, headache, metallic taste, exacerbation of asthma, VT proarrhythmia | ↓ Digoxin dose by 25%-50%; cimetidine and quinidine increase propafenone concentrations |
| Max. dose 300 mg PO q8h | Active metabolites accumulate in rapid metabolizers. Monitor QRS duration carefully | |||
| ↓ Dose if QRS prolonged >20% from baseline | ||||
| Class II | ||||
| Atenolol | 50-200 mg PO daily | Caution in patients with CHF or bronchospastic lung disease | Bradycardia, hypotension, dyspnea, fatigue, depression | With digoxin, Ca++ channel blockers, amiodarone |
| Monitor carefully in diabetic patients | ↓ Dose 25%-50% | |||
| ↓ Dose in moderate to severe renal insufficiency | Hypoglycemic agents | |||
| Bisoprolol | 2.5-10 mg PO bid | As per atenolol | As per atenolol | As per atenolol |
| Carvedilol | 3.25-50 mg PO bid | As per atenolol | As per atenolol | As per atenolol |
| Titrate dose every 1-2 weeks to achieve maximum tolerated dose | ||||
| yMetoprolol | IV: 5-15 mg | As per atenolol | As per atenolol | As per atenolol |
| 25-400 mg PO bid | ||||
| Nadolol | 20-160 mg PO daily | As per atenolol and ↓ dose in moderate to severe renal insufficiency | As per atenolol | As per atenolol |
| Propranolol | IV: 1-2 mg q2-4min | As per atenolol | As per atenolol | As per atenolol |
| prn × 4-5 doses | ||||
| 20-80 mg PO bid-tid | ||||
| Class III | ||||
| Amiodarone | For AF: 200 mg PO tid×2 week then 200 mg daily | Avoid high loading dose in setting of sinus bradycardia (HR < 50 beats/min) | pulmonary toxicity, CNS effects, hyper-/hypothyroidism, photosensitivity, corneal deposits, hepatic toxicity | ↓ Quinidine/procainamide dose by 50% |
| For VT: accelerated loading dose in hospital 400 mg PO tid×10-14 days, then 400 mg PO bid×7 days, then 300-400 mg PO daily | ↓ Digoxin dose by 50% | |||
| ↓ β-Blockers dose by 50% | ||||
| ↓ Warfarin dose by 50% | ||||
| Amiodarone IV | V: 150-300 mg over 20-30 min, then 0.5-1 mg/min; repeat boluses may be required | May cause hypotension | ||
| Dofetilide | 125-500 μg PO bid | ↓ Dose if QT interval prolongs after first dose by 15%; discontinue if QTc ≥ 550 ms | Headache; torsade de pointes VT | Cimetidine, verapamil, ketoconazole, trimethoprim alone or in combination with sulfamethoxazole |
| Sotalol | 80 mg PO q12h | ↓ Initial dose in renal failure | Torsade de pointes VT, hypotension, bradycardia, wheezing. Caution in CHF and bronchospastic lung disease | Digoxin/verapamil/other β-blockers may cause AV block, bradycardia |
| ↑ By 80 mg increments if QTc < 460 ms | ↓ Initial dose to 40 mg PO q12h in the elderly | |||
| Max. dose 240 mg PO q12h | ||||
| ↓ Dose if QTc ≥ 500 ms; discontinue if QTc ≥ 550 ms | ||||
| Dronedarone | 400 mg PO q12h | Diarrhea, increase in serum creatinine (inhibition of tubular transport), QTc prolongation | Digoxin/verapamil/other β-blockers may cause AV block. Bradycardia | |
| Class IV | ||||
| Diltiazem | IV: 0.25-0.35 mg/kg | Caution in patients with CHF | Bradycardia, hypotension, peripheral edema | β-Blockers, digoxin and amiodarone |
| 120-480 mg PO daily-bid | ||||
| Verapamil | IV: 5-15 mg | Caution in patients with CHF | Bradycardia, hypotension, constipation, flushing | β-Blockers, digoxin, amiodarone |
| 80 mg PO tid; max. dose 120 mg qid to 240 mg bid | ||||
| Other | ||||
| Digoxin | 0.0625-0.25 mg PO daily | ↓ Dose in renal failure | Arrhythmias, visual disturbance, nausea, vomiting | β-Blockers; calcium channel blockers, quinidine, propafenone, procainamide, amiodarone |
| Digoxin IV | IV: 0.25-1.0 mg over 20-30 minutes | |||
VENTRICULAR TACHYARRHYTHMIAS
Sustained ventricular arrhythmias including monomorphic VT, polymorphic VT or ventricular fibrillation (VF) usually occur in the setting of structural heart disease—most frequently in the setting of coronary artery disease, previous myocardial infarction and poor left ventricular function.20 However, any form of structural heart disease may be associated with ventricular arrhythmias. As well, some individuals have primary electrical disease usually associated with a mutation affecting one or more ion channels or proteins that regulate ion channels, for example, long QT syndrome, Brugada syndrome.21
The QRS complexes are uniform in monomorphic VT, whereas the QRS complexes are continuously varying in polymorphic VT. In VF, the surface ECG is disorganized without discernible QRS complexes. VT may precede the development of VF particularly in patients with a prior history of myocardial infarction. These arrhythmias are considered to be sustained if they last longer than 30 seconds or if they require acute intervention for termination.15,22
Multiple mechanism(s) contribute to VT. Monomorphic VT that develops in patients with a prior myocardial infarction is due to reentry near the border of the scar. In patients with dilated cardiomyopathy and an underlying intraventricular conduction delay, monomorphic VT usually with a left bundle branch block pattern may develop due to bundle branch reentry. In patients without structural heart disease, catecholamine-sensitive VT may originate in the right ventricular outflow tract due to triggered activity initiated via cyclic AMP. A verapamil-sensitive monomorphic VT that originates in the region of the left posterior fascicle is thought to be due to triggered activity.21 Such VTs that occur in patients with no structural heart disease (or channelopathy) are usually not life threatening.
Polymorphic VT in the setting of a normal QT interval usually occurs in the setting of acute ischemia or significant hemodynamic instability, although it may also occur in otherwise healthy individuals due to a mutation of the ryanodine receptor.23,24 Torsade de pointes VT is a polymorphic, pause-dependent VT that develops in association with drugs or pathophysiologic conditions which excessively prolong the QT interval (Fig. 36-2
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


