Calcium and Phosphorus Metabolism
Robert J. Cunningham III
This chapter discusses the complex systems that control the homeostasis of calcium and phosphorus. The aim is to provide an understanding of the relationships between vitamin D, parathyroid hormone (PTH), calcium, and phosphorus. Diseases that disturb the balance of this system are also discussed.
Calcium ions are critical for normal neuromuscular function and must be maintained at very precise concentrations to allow normal function. The two systems that control calcium metabolism are schematically outlined in Figure 18.1. The system responsible for the long-term control of calcium metabolism is the vitamin D pathway. Vitamin D3 (cholecalciferol) is formed in the skin on exposure to ultraviolet light. It is then transported to the liver, where it undergoes chemical conversion to 25-D3 (25-hydroxy-cholecalciferol). 25-D3 in turn circulates to the renal cortex, where it is converted by the addition of a second hydroxyl group to 1,25-D3 (1,25-dihydroxycholecalciferol). This compound is transported to the cells of the small intestine, stimulates protein synthesis, and ultimately leads to an increase in calcium absorption from the gastrointestinal tract. The entire process requires time, approximately 24 hours, because proteins must be synthesized in the intestinal cells in response to the stimulus provided by 1, 25-D3.
PTH provides a rapid response system that allows a nearly instantaneous increase in ionized calcium (Fig. 18.1, upper right panel). When the serum level of ionized calcium decreases, the parathyroid gland is stimulated to produce and release PTH. The released PTH mobilizes calcium directly from bone, so that the serum level of calcium increases immediately. PTH also induces an increase
in 1α-hydroxylase activity by stimulating the transcription of RNA. This increases the production of 1,25-D3 and hence increases the absorption of calcium which feeds back to turn off the secretion of PTH.
in 1α-hydroxylase activity by stimulating the transcription of RNA. This increases the production of 1,25-D3 and hence increases the absorption of calcium which feeds back to turn off the secretion of PTH.
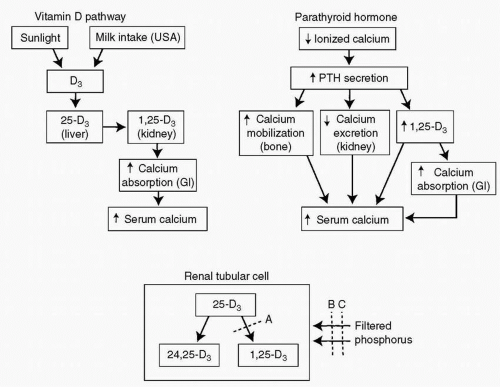 Figure 18.1 The two systems that control calcium and phosphorus metabolism are the vitamin D pathway (upper left panel) and the parathyroid hormone (PTH) (upper right panel). The renal tubular cell is the site of 1 α-hydroxylation of 25-D3 and also the site of phosphorus reabsorption. Line A represents the defect that occurs in vitamin D-dependency rickets. Line B represents the block in the transport of filtered phosphate into the renal tubular cell that occurs in vitamin D-resistance rickets. Line C represents the decrease in phosphate reabsorption that is caused by PTH. GI, gastrointestinal. |
Another effect of PTH is to cause a decrease in renal phosphate reabsorption so that there is some phosphate wasting in patients who have long-term elevated PTH levels.
VITAMIN D-DEFICIENCY RICKETS
A number of factors predispose to the development of vitamin D deficiency:
Lack of exposure to sunlight
Lack of intake of commercial milk products
Anticonvulsant therapy (particularly with phenobarbital or phenytoin)
Intestinal malabsorption syndromes, particularly those that cause fat malabsorption (e.g., cystic fibrosis)
Exposure to sunlight results in the formation of vitamin D3 in the skin cells; therefore, children with significant exposure to sunlight have adequate levels of vitamin D3 and rarely have vitamin D-deficiency rickets. Therefore, deficiency rickets is seen more often in the northern climates; in patients who, for cultural or religious reasons, have most of their skin surface covered with clothing; and in individuals with darker skin. In Siberia, school children are stripped to their underwear and made to stand in front of an ultraviolet light for 30 minutes three times a week during the winter. In this way, they receive adequate exposure to ultraviolet light to prevent vitamin D deficiency. By law, commercial milk products sold in the United States are fortified with vitamin D, and children who consume commercial milk products are unlikely to have vitamin D deficiency. Patients who consume skim milk are at greater risk of developing vitamin D deficiency, not because the milk is unfortified, but because the lower fat content of skim milk causes a lower absorption than milk with a higher fat content. Breast-fed infants are also at higher risk, particularly if the mother has a low intake of vitamin D, is dark skinned, or has limited exposure to sunlight. Anticonvulsant therapy does not influence the synthesis of vitamin D, but rather stimulates the P-450 system in the liver and accelerates the catabolism of 1, 25-D3; this accelerated breakdown may result in deficiency rickets. Vitamin D is fat-soluble; therefore, vitamin D deficiency may develop in patients who depend on an oral intake of vitamin D and have a fat malabsorption syndrome.
Rickets develops in stages. The stage I is that, as a consequence of vitamin D3 deficiency, there is insufficient absorption of calcium from the diet, and with time, the serum calcium levels fall. The fall leads to the synthesis and release of PTH and the mobilization of calcium from bone. In stage II, phosphorus reabsorption is decreased, and the renal tubular concentration of phosphate decreases. However, no 25-D3 is presented to the renal tubular cells, so that no conversion of 25-D3 to 1,25-D3 and no increase in calcium absorption takes place. Therefore, calcium levels are maintained only by the continued release of PTH, and the mobilization of calcium from bone continues. As the disease progresses to stage III, the serum calcium levels fall, and the calcification of bony matrix becomes inadequate. The
product of calcium and phosphorus (calcium × phosphorus) must be higher than 35 to 40 mg/dL for normal mineralization of bone to proceed. The calcium and phosphorus precipitate into a cartilaginous matrix; if the concentration of either is inadequate, precipitation may not occur.
product of calcium and phosphorus (calcium × phosphorus) must be higher than 35 to 40 mg/dL for normal mineralization of bone to proceed. The calcium and phosphorus precipitate into a cartilaginous matrix; if the concentration of either is inadequate, precipitation may not occur.
The clinical signs and symptoms of vitamin D deficiency include:
Tetany
Growth retardation
Frontal bossing
Rachitic rosary
Widening of the wrists or knee joints
Subluxation of the wrists, knees, or ankles at the epiphyseal plates
Seizures
Seizures result from hypocalcemia and are a common presenting symptom in infants and often occur in the springtime. Vitamin D deficiency develops during the winter with a lack of sunlight exposure and then in the springtime, vitamin D is synthesized as the infant is exposed to sunlight. This triggers an avid deposition of calcium into bone and this may happen so rapidly that the ionized calcium in the serum falls with resulting hypocalcemic seizures. Tetany may also result from hypocalcemia but is less often a presenting symptom. Growth retardation results from poor bone mineralization and the failure of unmineralized bone to grow. The frontal bossing, rachitic rosary, and widening of the wrist or knee joints all have the same underlying cause. The cartilaginous matrix proliferates, and this tissue mounds up on itself, causing the formation of lumps at the costochondral junctions (the rosary), the frontal bossing, and the widening of the wrists and knees. The cartilaginous matrix is not calcified and therefore has little compressive strength. It is susceptible to trauma, and bending or subluxation may occur during minor trauma, which is why rachitic patients may have bent joints.
The radiologic findings correspond to the clinical findings and include widening of the epiphyses and fraying at the epiphyseal junctions (Fig. 18.2). The bony metaphyses may also be widened, the long bones may have a ball of matrix at the growing ends, and fractures may be visualized. Bone mineralization does not proceed normally and PTH breaks down bone that has already formed. Both processes predispose to fractures.
Laboratory findings in vitamin D-deficiency rickets include:
Low serum level of calcium
Low serum level of phosphorus
High level of alkaline phosphatase
High serum level of PTH
Low serum level of 25-D3
The treatment of vitamin D-deficiency rickets is the administration of vitamin D3, which provides a substrate for the formation of 1,25-D3 and allows an equilibration of the calcium, phosphorus, 1,25-D3, and PTH levels.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


