In humans, using nuclear magnetic resonance spectroscopy (MRS), lactate contribution to brain energy metabolism was reported to be less than 10% under basal conditions. However, this increased to as much as 60% at elevated plasma lactate levels [6]. Recent meta-analysis indicated that lactate supplements glucose and supports an increase in cerebral energy metabolism during increased physical activity in humans [7].
C. Clinical measures of cerebral oxygenation and metabolism
1. Jugular venous oximetry (see Chapter 28). A catheter is inserted into an internal jugular vein and advanced to the jugular bulb. The right side is often used because it is usually dominant. Jugular venous oxygen saturation (SjvO2) gives information about the balance between global brain oxygen delivery and metabolic demand. The normal SjvO2 is 55% to 75%. The normal lower value of SjvO2 compared to cardiac mixed venous saturation reflects high oxygen metabolism in the brain. SjvO2 levels less than 55% suggest that the cerebral oxygen supply is inadequate to meet the metabolic demand, while a level higher than 80% indicates relative hyperemia. The arterial to jugular venous oxygen concentration difference (AjvdO2) has been used to estimate brain metabolism. The normal AjvdO2 is 4- to 8-mL O2/100-mL blood.
2. Brain tissue oxygen tension (see Chapter 28). Measurement of brain tissue oxygen tension (PtO2) using an oxygen-sensing electrode provides information regarding focal brain tissue oxygenation; although in doing so, global changes might be missed. Brain PtO2 is related to other physiologic variables. It is increased by an increase in the inspired oxygen tension, arterial blood oxygen tension (PaO2), red blood cell concentration, and mean arterial pressure (MAP) or cerebral perfusion pressure (CPP). There is no established critical level of brain PtO2. Normal brain PtO2 values are in the range of 35 to 50 mm Hg.
3. Near infrared spectroscopy (see Chapter 28). This noninvasive measurement is based on the transmission and absorption of near infrared light (700 to 950 nm) as it passes through tissue. Oxygenated and deoxygenated hemoglobin have different absorption spectra and brain oxygenation can be calculated by their relative absorption of near infrared light. Near infrared spectroscopy (NIRS) interrogates all tissue within the field of view. Thus, NIRS measurements of hemoglobin saturation reflect influences from arterial, venous, and capillary blood [8]. Currently, there is no standard among commercial NIRS systems; although most provide an absolute measure of brain tissue oxygen saturation in some form and display this as a simple percentage value. The recent development of NIRS-derived measurements of cytochrome c oxidase has been validated in animal models as a measurement of cellular status. It offers a potential to assess intramitochondrial respiration in traumatic brain injury.
4. Cerebral microdialysis. This technique measures cellular functions through accumulation of substrates or metabolic by-products by analyzing microdialysate concentrations of glucose, lactate, pyruvate, glycerol, and/or glutamate. Cerebral ischemia or hypoxia is associated with marked increases in the lactate/pyruvate ratio and glycerol and glutamate levels [9].
CLINICAL PEARL
• Critical brain tissue pO2 is 15 to 20 mm Hg.
• Brain consumes glucose as its preferred substrate during normal resting conditions.
• Lactate supplements glucose as an energy source and is increasingly used by the brain during neuronal activation (e.g., exercise, severe hypoglycemia, and hypoxia).
II. Cerebral blood flow
2
A. General concepts. Tight regulation of CBF is important to meet the energy demands of active neurons. This process is often labeled “neurovascular coupling.” Total CBF at rest in humans is about 800 mL/min (50 mL/100 g/min), which is 15% to 20% of the cardiac output. In normal physiologic states, total blood flow to the brain is remarkably constant. This is due in part to the prominent contributions of both large arteries and parenchymal arterioles to overall vascular resistance.
Neuronal function and cellular integrity can be compromised by reductions in regional cerebral blood flow (rCBF). During prolonged focal ischemia, brain tissue may develop a localized injury pattern consisting of a core of tissue destined for destruction (infarction) surrounded by a “penumbra” of metabolically semistable tissue that has the potential for full recovery, or may contribute to expansion of the zone of infarction. Initially, penumbra was proposed to be related to an area with 50% reduction in evoked potential amplitude. An increased extracellular K+ concentration, which is linked to energy failure, contributes to cell death in the infarct core. The spread of the elevated K+ into the penumbral zone can place further demands on energy-generating processes within penumbral cells and recruit more tissue into the infarct core. The use of PET imaging has permitted measurements of rCBF, CMRO2, rCMRglu, and oxygen extraction fraction (OEF) in penumbra and infarction regions. In humans, penumbra is defined as the area where rCBF decreases to 12 to 22 mL/100 g/min, rCMRO2 remains above 65 μmol/100 g/min, and OEF is increased to more than 50%. Infarction usually corresponds to rCBF below 12 mL/100 g/min and rCMRO2 below 65 μm/100 g/min [10].
B. Regulation of CBF
1. Myogenic response. The myogenic response is the intrinsic property of vascular smooth muscle to react to changes in mechanical input or intravascular pressure. The smooth muscle of large cerebral arteries and small arterioles dilate in response to decreased pressure and constrict in response to increases in pressure, therefore contributing to autoregulation of blood flow. Local metabolites and release of vasoactive factors from endothelium and perivascular nerves can influence myogenic tone and vascular resistance. The myogenic response mechanism involves two processes: Myogenic tone and myogenic reactivity. An excessively high arterial pressure (above the autoregulatory range) results in a condition known as forced dilation, which involves a marked increase in vessel diameter and loss of tone.
Initiation of the myogenic constrictor response occurs through ionic and enzymatic processes that lead to accumulation of intracellular calcium. Increased pressure causes depolarization of smooth muscle cell membrane promoting calcium influx via opening of voltage-operated calcium channels (Cav). Wall tension is proposed as a stimulus for an initiation of the myogenic response. Stretch-activated cation channels (i.e., transient receptor potential [TRP] channels) are thought to be the sensor. The increase in intracellular calcium leads to myosin light chain phosphorylation and promotes vasoconstriction. Other signaling mechanisms include activation of protein kinase C (PKC) and RhoA-Rho kinase pathways.
A feedback loop exists to prevent excessive myogenic constriction. This mechanism, which limits constriction-induced depolarization, is the activation of calcium-activated potassium channels (especially large-conductance BKca channels) expressed on cerebral artery smooth muscle. Activation of BKca channels causes hyperpolarization and attenuation of myogenic vasoconstrictive responses [11].
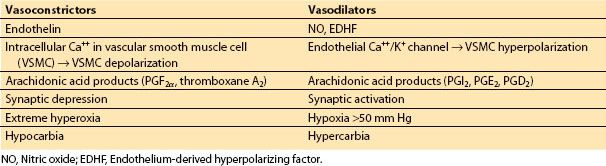
2. Endothelial contributions. Cerebral endothelial cells form tight junctions, establishing the blood–brain barrier. Endothelial dysfunction has been proposed to be an important factor in the pathogenesis of cardiovascular disease such as atherosclerosis, stroke, diabetes, and subarachnoid hemorrhage. The endothelium produces several vasoactive substances that can have significant influence on vascular tone: Nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), prostacyclin, and other eicosanoids.
a. Nitric oxide. Nitric oxide synthase (NOS) is the enzyme responsible for the O2-dependent conversion of L-arginine to NO. There are three isoforms of NOS. Neuronal (nNOS or NOS1) is constitutively expressed in neurons under normal conditions. Inducible (iNOS or NOS2) is not normally expressed in the brain but can be induced under pathologic conditions. Lastly, endothelial (eNOS, NOS3) is abundantly expressed in the endothelium. NO generated by eNOS diffuses to vascular smooth muscle where it binds to and activates soluble guanylate cyclase, resulting in increased levels of cyclic guanine monophosphate (cGMP) and activation of protein kinase G (PKG), which causes vasodilation by opening K+ channels (BKca) and/or reducing the sensitivity of the contractile machinery to Ca+. eNOS is regulated by calcium, heat shock protein (HSP90), serine/threonine phosphorylation, and tyrosine phosphorylation.
The NO system is altered in many pathologic states. Under conditions of limited substrate or low levels of the cofactor tetrahydrobiopterin (BH4), eNOS can become uncoupled from NO production leading to the production of superoxide (O2−) [12]. This has been implicated in brain damage arising from atherosclerosis, diabetes, and hypertension.
CLINICAL PEARL
Statins (HMG-CoA reductase blockers) improve outcome in stroke via increased eNOS expression and reduced NADPH oxidase activity. This action on eNOS involves preservation of eNOS mRNA and is unrelated to its cholesterol-lowering effect.
b. Endothelium-derived hyperpolarizing factor. EDHF-mediated dilation occurs in the presence of NOS and cyclooxygenase (COX) metabolite inhibition. The EDHF response requires increased endothelial calcium and activation of small- and intermediate-conductance calcium-activated potassium channels (SKCa and IKCa). Endothelial hyperpolarization then transfers to vascular smooth muscle hyperpolarization and vasodilation; the mechanism is unclear, but may involve gap junctions [13]. Emerging evidence supports a role for EDHF as a contributing factor/mediator to resting tone in small cerebral arterioles, thus influencing resting CBF. In addition, the EDHF-mediated dilating system is upregulated after stroke and traumatic brain injury, whereas endothelium-derived NO dilating system is decreased [12]. This suggests a role for EDHF as a compensatory vasodilating mechanism when NO contributions are diminished during pathologic conditions.
c. Prostacyclin and other eicosanoids. In endothelial cells and astrocytes, arachidonic acid is metabolized to vasoactive substances that are either vasoconstricting or vasodilating in nature. These products are collectively called eicosanoids. Arachidonic acid, once liberated from phospholipid membrane (via the action of phospholipase A2) is further metabolized by COX, lipoxygenase, epoxygenase, or hydroxylase. The COX pathways have been well studied. The two principal COX isoforms are variably expressed in neurons, glial cells, and cerebral endothelial cells. The products of COX pathway can be vasodilating such as prostacyclin (PGI2), prostaglandin E2, and prostagladin D2, or vasoconstricting such as prostaglandin F2a and thromboxane A2. PGI2 is most studied in cerebral endothelium cell. PGI2 diffuses to the smooth muscle where it binds to G-protein–coupled receptor and activates adenylate cyclase and protein kinase A (PKA). The increase in PKA activity can promote the opening of certain K+ channels (BKca; inward-rectifier [Kir]) producing smooth muscle hyperpolarization and voltage-sensitive Ca2+ channel closure, resulting in decreased intracellular Ca2+ levels and vasodilation. The epoxygenase and hydroxylase pathways yield vasodilating and vasoconstricting products, respectively through actions toward BKca channels (Table 1.1).
Table 1.1 Some factors influencing cerebrovascular tone
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


