Arrhythmias, Pacing, and Cardioversion
KEY POINTS
1 Many arrhythmias require drug treatment; those that are asymptomatic, chronic, and stable, or related to a temporary physiological disturbance (e.g., transient hypoxemia, electrolyte abnormalities) can be observed while the underlying problems are corrected.
2 Correction of hypoxemia and electrolyte disturbances is essential to minimize the risk of arrhythmias and to facilitate conversion to a stable baseline rhythm.
3 Symptomatic patients with tachyarrhythmias of uncertain origin should usually be treated as if they have ventricular arrhythmias, regardless of QRS complex width, especially if hypotensive.
4 Narrow complex tachycardias can be diagnosed or terminated in most cases by using carotid sinus massage and intravenous adenosine.
5 A large fraction of patients treated with antiarrhythmics will develop side effects, which may include new or worsened arrhythmias.
6 Temporary pacemakers are indicated for: (a) high-grade (especially symptomatic) atrioventricular block following myocardial infarctino, (b) overdrive suppression of refractory atrial tachyarrhythmias, (c) suppression of torsades de pointes resulting from bradycardia, (d) sick sinus syndrome, and (e) control of postcardiac surgery arrhythmias.
7 Standby pacing capability should be available during high-risk cardioversions and during right heart catheterization in patients with underlying conduction system disease (e.g., left bundle branch block).
The treatment of arrhythmias has become less common and much simpler over the last two decades as a result of a series of important discoveries. For example, it was revealed that it is unnecessary to convert most patients with atrial fibrillation (A-fib) to sinus rhythm and the suppression of premature ventricular contractions (PVCs) does not improve outcomes. In addition, many of the drugs used for years to control ventricular arrhythmias were abandoned because they were found to increase mortality. The sophistication and effectiveness of implantable and transthoracic pacing systems dramatically improved. Radiofrequency ablation made many chronic troublesome arrhythmias curable, and for arrhythmias that cannot be cured, (e.g., genetic long QT syndrome, recurrent sudden death), implantable defibrillators have been lifesaving. Adenosine has been immensely helpful for diagnosis and therapy, and the role of amiodarone in the treatment was refined. Finally, the widespread application of rapid reperfusion strategies for acute coronary syndrome eliminated millions of periinfarction arrhythmias and probably as many arrhythmias from postinfarction heart failure. Despite these advances, the critical care physician must still be able to make an accurate diagnosis of an arrhythmia by rapidly interpreting an electrocardiogram (ECG) and provide appropriate emergency treatment.
▪ COMPONENTS OF THE ELECTROCARDIOGRAM
The first step in evaluation of the ECG is to identify atrial activity (P waves) which is best seen in the inferior leads (II, III, and aVF). P wave shape should be examined for consistency; the pattern should be studied for evidence of atrial flutter or fibrillation and for the position and consistency of the P wave relative to the QRS complex. P wave inversion in limb lead II signifies retrograde atrial depolarization diagnostic of a nonsinus mechanism. After the atrial rhythm has been characterized, ventricular activity
(QRS complex) should be examined. If the QRS is narrow, ventricular depolarization most likely occurs in response to normal sequential atrioventricular (AV) conduction or at least is of supraventricular origin. A QRS complex (>0.12 s) suggests (a) ventricular origin, (b) an aberrantly conducted supraventricular beat, (c) a bypass pathway, or (d) supraventricular conduction delayed by a drug (e.g., tricyclic antidepressant) – electrolyte (e.g., hyperkalemia) abnormality. The QRS should be examined for regularity, rate, and the relationship to the P waves. If every QRS complex is not preceded by a P wave, some form of AV block, or A-fib or flutter, or ventricular tachycardia (VT), is likely. Because of the normal delays associated with AV nodal conduction, a QRS complex occurring less than 0.1 s after a P wave is unlikely to be related to it.
(QRS complex) should be examined. If the QRS is narrow, ventricular depolarization most likely occurs in response to normal sequential atrioventricular (AV) conduction or at least is of supraventricular origin. A QRS complex (>0.12 s) suggests (a) ventricular origin, (b) an aberrantly conducted supraventricular beat, (c) a bypass pathway, or (d) supraventricular conduction delayed by a drug (e.g., tricyclic antidepressant) – electrolyte (e.g., hyperkalemia) abnormality. The QRS should be examined for regularity, rate, and the relationship to the P waves. If every QRS complex is not preceded by a P wave, some form of AV block, or A-fib or flutter, or ventricular tachycardia (VT), is likely. Because of the normal delays associated with AV nodal conduction, a QRS complex occurring less than 0.1 s after a P wave is unlikely to be related to it.
▪ GENERAL APPROACH TO ARRHYTHMIAS
Acute arrhythmias are detrimental when they are symptomatic, reduce tissue perfusion, or increase myocardial oxygen demand. Chronic tachyarrhythmias can also be harmful by causing cardiomyopathy. In making management decisions, the patient’s symptoms; adequacy of perfusion; the risks of treatment versus observation; and chronicity of the problem must be considered. Tachyarrhythmias evoking unconsciousness, hypotension, pulmonary edema, or angina should be terminated immediately as should symptomatic bradycardia. Patients with isolated PVCs lacking evidence of heart failure or ischemia have an excellent prognosis without treatment. In such patients, drug suppression of the arrhythmia is unlikely to improve outcome, but is apt to produce untoward side effects. A past history of well-tolerated arrhythmia similar to the one currently present also suggests that rapid treatment is not necessary. Conversely, patients with myocardial ischemia and those with a history of malignant or degenerative arrhythmias should be treated aggressively. Arrhythmias are often exacerbated if not provoked by electrolyte disturbances, mechanical irritation of the heart, drugs, and ischemia. Thus, hypokalemia or hyperkalemia, hypomagnesemia, alkalosis, anemia, and hypoxemia all exacerbate arrhythmic tendencies. Intracardiac catheters, pacemaker malfunction, digitalis, theophylline, and sympathomimetic agents (e.g., catecholamines, cocaine) can provoke a wide variety of arrhythmias that cease upon their removal. It is also clear that several antiarrhythmic drugs (e.g., quinidine, sotalol, flecainide) can have serious proarrhythmic effects. Electrical instability is also heightened by ischemia. For example, hypotension reduces myocardial perfusion, whereas excess intravascular volume or high ventricular afterload can increase wall tension and oxygen demand.
Dealing with Uncertainty
Asymptomatic, or minimally symptomatic narrow complex tachyarrhythmias, and pulseless arrhythmias rarely present diagnostic or therapeutic dilemmas. By contrast, an unfamiliar arrhythmia, especially a wide complex tachycardia (WCT), in a patient with a moderate decrease in blood pressure or modest symptoms is often anxiety provoking. The first step when confronted with an unfamiliar arrhythmia is to confirm that it is real. Electrical artifacts may occur as a result of poor surface electrode contact or electromechanical devices such as aortic balloon or infusion pumps. Shivering, seizure activity, and tremors of Parkinson disease can produce ECG artifacts that may be confused with serious arrhythmias.
The most consternation is caused by monomorphic WCT not clearly of ventricular or supraventricular origin. To avoid mistakes under pressure, it is important to develop an approach to diagnosis and therapy in advance (Table 4-1). When patients are hemodynamically compromised, it is best to treat arrhythmias as if they were life threatening and the vast majority of WCTs are ventricular.
However, it is important to exclude the presence of high-grade AV block. (Infranodal escape rhythms must not be terminated before treating the underlying heart block.) Hence, patients with WCT in distress should receive either cardioversion or drug therapy for VT (e.g., amiodarone, lidocaine, procainamide) depending on clinical urgency. Traditionally, lidocaine was the drug of first choice, and failure to respond to it supports a diagnosis of supraventricular tachycardia (SVT) with aberrant conduction. (In this setting, procainamide and amiodarone are good choices because they will control many types of SVT and VT.) Although these drugs rarely help clarify the diagnosis, they often control the rhythm long enough to get expert advice or perform more sophisticated diagnostic maneuvers. In the patient failing a trial of lidocaine, adenosine may also be tried. (By transiently blocking the AV node, adenosine is very effective at slowing or terminating SVT.) In cases of WCT, verapamil or diltiazem is a suboptimal choice for empirical therapy because their cardiodepressant and vasodilating properties often further lower the blood pressure in the setting of VT, and SVTs utilizing a bypass tract can be accelerated. Similarly, adenosine is not a good choice if the patient is suspected to have a bypass tract. A discussion of the most common arrhythmias and their treatment follows.
However, it is important to exclude the presence of high-grade AV block. (Infranodal escape rhythms must not be terminated before treating the underlying heart block.) Hence, patients with WCT in distress should receive either cardioversion or drug therapy for VT (e.g., amiodarone, lidocaine, procainamide) depending on clinical urgency. Traditionally, lidocaine was the drug of first choice, and failure to respond to it supports a diagnosis of supraventricular tachycardia (SVT) with aberrant conduction. (In this setting, procainamide and amiodarone are good choices because they will control many types of SVT and VT.) Although these drugs rarely help clarify the diagnosis, they often control the rhythm long enough to get expert advice or perform more sophisticated diagnostic maneuvers. In the patient failing a trial of lidocaine, adenosine may also be tried. (By transiently blocking the AV node, adenosine is very effective at slowing or terminating SVT.) In cases of WCT, verapamil or diltiazem is a suboptimal choice for empirical therapy because their cardiodepressant and vasodilating properties often further lower the blood pressure in the setting of VT, and SVTs utilizing a bypass tract can be accelerated. Similarly, adenosine is not a good choice if the patient is suspected to have a bypass tract. A discussion of the most common arrhythmias and their treatment follows.
TABLE 4-1 TREATMENT FOR REGULAR MONOMORPHIC WIDE COMPLEX TACHYCARDIA OF UNCERTAIN ORIGIN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
▪ TACHYARRHYTHMIAS
Sinus Tachycardia
Sinus tachycardia (ST) is the primary means of raising cardiac output in response to metabolic demands; thus, it is physiologic in the setting of exercise, fever, or hyperthyroidism. ST is also appropriate compensation for hypovolemia, limited stroke volume, reduced systemic vascular resistance, or reduced myocardial compliance. Anxiety, pain, and drugs (e.g., catecholamines, cocaine, theophylline) may also be responsible. Unless ST causes ischemia by increasing myocardial oxygen consumption, or precipitates pulmonary edema by shortening diastolic filling time in a patient with reduced ventricular compliance, it is simply a marker of illness. The best therapy for ST is to treat the underlying cause. In patients with symptomatic ischemia, β-blockade often proves helpful. However, β-blockers should be used cautiously in tachycardic patients with hypotension, acute infarction, or chronic congestive heart failure because ST often reflects incipient decompensation. Likewise, caution is indicated using β-blocker in patients with obstructive lung disease because of the risk of exacerbating bronchospasm.
Nonsinus Supraventricular Tachycardias
The nomenclature surrounding SVT is confusing but the concepts are simple. SVT usually results from a self-perpetuating reentry mechanism; much less commonly, SVT stems from rapid discharge of an ectopic atrial focus. Graphic examples of the most common forms of SVT are shown in Figure 4.1.
Reentrant Tachycardias Involving the AV Node
Reentrant SVTs occur when two potential transmission pathways have differing conduction speeds and refractory periods permitting a reverberating circuit to develop. This group of arrhythmias is classified by whether that circuit lies solely within the AV node or whether one limb of the circuit bypasses the AV node. By far the most common form is AV nodal reentrant tachycardia (AVNRT) in which a “micro-reentrant” pathway exists entirely
within the AV node. Although sometimes confused with atrial flutter with 2:1 conduction, AVNRT can usually be identified by its isoelectric inter-QRS baseline and slightly irregular, narrow QRS, occurring at a rate 150 to 200 beats/min. QRS complexes frequently exhibit a rate-related, right-bundle branch block pattern that may simulate VT. (P waves are often buried in the QRS complex or T wave, producing a “pseudo-S wave.”) When visible, P waves are frequently inverted in the inferior leads because atrial depolarization characteristically begins in the AV node located low in the right atrium and spreads cephalad. Unlike A-fib or flutter, in which the ventricular response slows to vagal stimulation or adenosine therapy, AVNRT either remains completely unaffected or stops abruptly.
within the AV node. Although sometimes confused with atrial flutter with 2:1 conduction, AVNRT can usually be identified by its isoelectric inter-QRS baseline and slightly irregular, narrow QRS, occurring at a rate 150 to 200 beats/min. QRS complexes frequently exhibit a rate-related, right-bundle branch block pattern that may simulate VT. (P waves are often buried in the QRS complex or T wave, producing a “pseudo-S wave.”) When visible, P waves are frequently inverted in the inferior leads because atrial depolarization characteristically begins in the AV node located low in the right atrium and spreads cephalad. Unlike A-fib or flutter, in which the ventricular response slows to vagal stimulation or adenosine therapy, AVNRT either remains completely unaffected or stops abruptly.
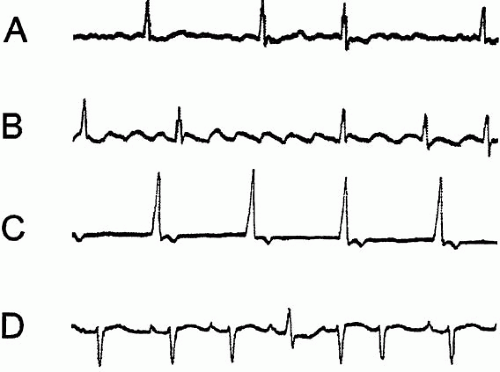 ▪ FIGURE 4-1 Stylized electrocardiographic tracings illustrating the distinguishing features of the most common supraventricular arrhythmias. Panel A shows the irregular ventricular response and absence of well-defined P waves characteristic of A-fib. By contrast, Panel B illustrates the rapid inverted “sawtooth” atrial depolarizations of atrial flutter. Commonly a 2:1 ventricular response often results in a ventricular rate of 150 beats/min. Panel C shows the most common pattern of AVNRT in which an isoelectric baseline is punctuated by slightly irregular narrow complex ventricular depolarizations. Close examination reveals inverted monomorphic P waves buried in the QRS and T waves. Panel D illustrates the characteristic polymorphic P wave pattern of MAT. |
The less common form of reentrant SVT is AV reentrant tachycardia (AVRT) in which ventricular rates are typically a bit faster (150 to 250 beats/min). AVRT is caused by a “macro-reentrant” circuit in which one conduction limb goes through the AV node and one limb bypasses it. AVRT is subclassified by the direction the current takes through the AV node. If antegrade conduction is through the AV node, it is termed “orthodromic.” Unless there are rate-related conduction delays, in orthodromic AVRT the QRS complex is of normal width because conduction follows the usual AV-node-His-Purkinje pathway. If antegrade conduction occurs through the accessory or bypass tract, the rhythm is termed “antidromic.” Antidromic AVRT may be identified (especially while the patient is in sinus rhythm) by a short PR interval, wider QRS, and delta waves indicative of ventricular preexcitation on the bypass tract. The rare, but best characterized preexcitation condition is Wolfe-Parkinson-White syndrome. Antidromic AVRT is important to recognize because such patients are at higher risk of sudden cardiac death, and it may respond paradoxically, sometimes catastrophically, to usual SVT treatments. Intra-SA node, or intra-atrial, reentrant rhythms may occur but will not be discussed further because they are rare.
Adenosine and vagal maneuvers like carotid sinus massage are valuable tools to distinguish reentrant SVTs from VT and from non-nodal-reentrant atrial arrhythmias like A-fib and flutter and ectopic atrial tachycardia. VT is unresponsive to vagal maneuvers, and if the rhythm is A-fib, only transient slowing of the ventricular rate is likely. If the rhythm is flutter, blocking the AV node will unmask the characteristic flutter waves but will not stop the atrial reentrant circuit.
SVTs are generally well tolerated and self-limited, often requiring no treatment with the possible exception of stopping exacerbating drugs (e.g., theophylline, catecholamines, and cocaine) and correcting electrolyte disorders. If treatment is needed, maneuvers or drugs that inhibit conduction through the AV node are highly effective. Massage of the nondominant carotid artery for 10 to 15 s, alone or in conjunction with the Valsalva maneuver, often interrupts AVNRT and AVRT. To avoid cerebral ischemia, both carotid arteries should not be compressed simultaneously, and vessels with bruits and those of patients with a history of stroke or transient ischemia should not be massaged. When mechanical maneuvers fail, drug intervention is indicated. Adenosine has supplanted calcium channel blockers (e.g., verapamil) as the initial therapy of choice for hemodynamically stable SVT. As a potent but short-lived AV node blocker, a 6 to 18 mg of IV (intravenously) adenosine dose terminates AVNRT and AVRT with a success rate equal to or greater than that of verapamil. Although very effective at stopping the original arrhythmia, up to 10% of patients with AVNRT and AVRT convert to A-fib. Adenosine does not terminate A-fib or flutter. Calcium channel blockers, like verapamil, represent second-line therapy but should be avoided when supraventricular origin is in doubt, hypotension is present, or a bypass tract is suspected. The nodal blocking effects may encourage atrial conduction over the bypass tract, and the vasodilating effects of calcium channel blockers may cause hypotension unless rhythm conversion occurs. If used, verapamil doses of 2.5 to 5 mg IV are usually adequate. (Comparable doses of diltiazem may be substituted, but nicardipine is less effective.) β-blockers like propranolol (0.5 to 1 mg every 5 min, up to a 4-mg total dose) or metoprolol may also be effective. If the patient’s ability to tolerate β-blockade is uncertain, the short-acting esmolol can be tried. Digoxin has long been used in the treatment of AVNRT but often requires hours for effect, making it most useful in hemodynamically stable patients and in those requiring prophylaxis. By boosting systemic blood pressure, vasoconstrictive drugs (e.g., phenylephrine) may reflexly decrease AV-nodal conduction. However, vasopressors may precipitate cardiac or cerebrovascular complications and are therefore usually avoided. Cholinergic stimulants like neostigmine can increase nodal vagal tone thereby ending nodal
reentrant arrhythmias but have an unacceptable side effect profile. Because all of the reentrant circuit is in the AV node in AVNRT, it tends to be easily broken by AV-nodal blocking measures. Because just one limb of the conducting circuit passes through the AV node in AVRT, adenosine (or any other AV-nodal blocker) often stops the arrhythmia but there is a risk; blocking antegrade conduction through the AV node may promote very rapid conduction through a bypass tract with a very rapid, even life-threatening (VT or VF) response. This complication is most likely in patients who develop A-fib or flutter with a bypass tract.
reentrant arrhythmias but have an unacceptable side effect profile. Because all of the reentrant circuit is in the AV node in AVNRT, it tends to be easily broken by AV-nodal blocking measures. Because just one limb of the conducting circuit passes through the AV node in AVRT, adenosine (or any other AV-nodal blocker) often stops the arrhythmia but there is a risk; blocking antegrade conduction through the AV node may promote very rapid conduction through a bypass tract with a very rapid, even life-threatening (VT or VF) response. This complication is most likely in patients who develop A-fib or flutter with a bypass tract.
Lidocaine has no effect on SVT. Type Ia antiarrhythmics (e.g., quinidine and procainamide) exert vagolytic effects and often worsen SVT by accelerating AV conduction unless nodal-blocking drugs are administered first. In refractory SVT, temporary overdrive atrial pacing may restore sinus rhythm and is easily done when an atrial pacer or pacing pulmonary artery catheter is already in place, as is common after cardiac surgery. Hemodynamically unstable SVT should be treated with low-energy (10 to 50 WS [watt-seconds]) synchronized cardioversion. An outline of therapy for SVT is presented in Table 4-2.
Primary Atrial Tachycardias
Ectopic Atrial Tachycardia
Ectopic atrial tachycardia can result from a single ectopic focus firing at a rapid rate, or more commonly from multiple rapidly discharging atrial foci. The later mechanism is known as multifocal atrial tachycardia (MAT) and most often occurs in association with obstructive lung disease or metabolic crisis, but it also complicates left ventricular failure, coronary artery disease, diabetes, sepsis, and toxicity with digitalis, theophylline, and sympathomimetic drugs. Among patients with lung disease, hypoxemia, hypercapnia, acidosis, alkalosis, pulmonary hypertension, and β-agonist therapy have all have been identified as risk factors. When multiple P-wave morphologies are conducted at a normal ventricular rate, the condition is referred to as a “wandering atrial pacemaker.” MAT is recognized by irregularly irregular QRS complexes of supraventricular origin, varying PR intervals, and the presence of at least three morphologically distinct P waveforms on an isoelectric baseline. (The less common unifocal atrial tachycardia has a single P wave morphology.) Comparable heart rates (100 to 180 beats/min) and beat-to-beat variation in PR and RR intervals often cause MAT to be confused with A-fib.
TABLE 4-2 TREATMENT PLAN FOR NARROW COMPLEX REGULAR TACHYCARDIA | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||

Because ectopic tachycardias do not depend on the AV node, measures to increase AV-nodal refractoriness are usually ineffective. Although β-blockers and calcium channel blockers may temporarily slow or convert MAT, the definitive treatment is to reverse the underlying cause. Correction of hypokalemia and supplementing magnesium, perhaps even if levels are within the normal range, can be helpful and is unlikely to be harmful unless renal insufficiency is present. (MgSO4 given as 2 gm IV may slow the rate, if not convert the rhythm.) Verapamil
(up to 20 mg), or diltiazem, can be useful by decreasing the frequency of the atrial impulses, not by blocking their entry to the ventricle. Unfortunately, verapamil commonly reduces blood pressure, an effect that to some extent can be ameliorated by pretreatment with calcium gluconate. β-blockade can also control the rate or less often abolish MAT, but has obvious limitations in a population of patients many of whom have lung disease. If β-blockers are used, short-acting agents (i.e., esmolol) or cardioselective blockers (i.e., metoprolol) make the most sense. Metoprolol, 5 mg IV every 10 min can be tried and if well tolerated, the patient can be converted to an oral dose of 50 to 100 mg once or twice daily. Neither cardioversion, digitalis, nor the antiarrhythmics lidocaine, quinidine, procainamide, and phenytoin benefit patients with MAT. The role of radiofrequency ablation for MAT is uncertain at this time. Whereas theophylline and β-agonists may occasionally precipitate MAT, their cautious use may improve underlying bronchospasm sufficiently to reverse the arrhythmia. In patients who demonstrate MAT in response to theophylline or β-agonists, corticosteroids or inhaled anticholinergics represent attractive alternative options for treating bronchospasm because they are not cardiostimulatory.
(up to 20 mg), or diltiazem, can be useful by decreasing the frequency of the atrial impulses, not by blocking their entry to the ventricle. Unfortunately, verapamil commonly reduces blood pressure, an effect that to some extent can be ameliorated by pretreatment with calcium gluconate. β-blockade can also control the rate or less often abolish MAT, but has obvious limitations in a population of patients many of whom have lung disease. If β-blockers are used, short-acting agents (i.e., esmolol) or cardioselective blockers (i.e., metoprolol) make the most sense. Metoprolol, 5 mg IV every 10 min can be tried and if well tolerated, the patient can be converted to an oral dose of 50 to 100 mg once or twice daily. Neither cardioversion, digitalis, nor the antiarrhythmics lidocaine, quinidine, procainamide, and phenytoin benefit patients with MAT. The role of radiofrequency ablation for MAT is uncertain at this time. Whereas theophylline and β-agonists may occasionally precipitate MAT, their cautious use may improve underlying bronchospasm sufficiently to reverse the arrhythmia. In patients who demonstrate MAT in response to theophylline or β-agonists, corticosteroids or inhaled anticholinergics represent attractive alternative options for treating bronchospasm because they are not cardiostimulatory.
Atrial Fibrillation
A-fib is a common, (prevalence approx. 5% among patients >70 years) chaotic atrial rhythm in which no single ectopic pacemaker captures the entire atrium, hence there is no detectable P wave on ECG. New onset A-fib often complicates chest surgery, pulmonary embolism, valvular heart disease, obstructive lung disease, and hyperthyroidism. The irregular ventricular rhythm may be confused with MAT, frequent premature atrial contractions, ST, or atrial flutter with variable AV block. Because there is no organized atrial depolarization or contraction to facilitate left ventricular priming, cardiac output may fall significantly, especially in patients with impaired ventricular compliance. Although the atria may depolarize up to 400 times/min, the AV node rarely conducts impulses at rates higher than 180 to 200 beats/min. However, fever, sepsis, vagolytic drugs, and the presence of accessory conduction pathways may increase the ventricular response. On physical examination, A-fib is suggested by a fluctuating S1 (because of varying mitral valve position at the onset of ventricular systole) and a pulse deficit (because of occasional systoles with low ejection volumes). There are three prominent risks of A-fib: hypoperfusion from too rapid a ventricular rate, systemic embolism from clot formation in the noncontractile atrium, and cardiomyopathy from chronic tachycardia.
Treatment is guided by ventricular rate, hemodynamic adequacy, baseline left ventricular function, duration of the rhythm, and presence of a nodal bypass tract. Acute hemodynamic compromise from a rapid ventricular rate (>150) mandates synchronized cardioversion (100 to 200 WS of monophasic energy or the biphasic equivalent). Occasionally, higher energy levels are required. (The longer the duration, the more resistant A-fib is to conversion.) Ventricular rates greater than 200 beats/min suggest accelerated conduction due to vagolytic drugs (e.g., type Ia antiarrhythmics) or presence of an accelerated conduction pathway. If the ventricular rate is less than 60 beats/min, drug effect (e.g., digitalis, β-blockers, calcium channel blockers) or conduction system disease should be suspected. In untreated patients with a slow ventricular response, electrical cardioversion, or nodal-blocking drugs may produce symptomatic bradycardia or even asystole; a risk that is sufficiently high that a temporary pacemaker should be inserted prior to attempting cardioversion.
There is no rush to correct chronic, hemodynamically stable A-fib. Before conversion is attempted, the likelihood of attaining and maintaining sinus rhythm should be assessed and the risk of systemic embolism should be considered. When left atrial diameter exceeds 4 cm, conversion to stable sinus rhythm is unlikely. Although many clinicians advocate at least one attempt at restoring sinus rhythm, most patients do quite well if anticoagulated and the resting ventricular rate is maintained less than 100 beats/min. Landmark trials indicate that restoration of sinus rhythm does not result in a significant reduction of the risk of embolization in the anticoagulated patient, but without anticoagulation the annual risk is substantially higher.
For hemodynamically stable A-fib, the first step in treatment is to slow the resting ventricular rate to ≤100 beats/min. If ventricular function is good, calcium channel or β-blockers are preferred rate-controlling agents. If ventricular function is impaired, digoxin is a better choice, although amiodarone and diltiazem can be used with caution. With digoxin alone or in combination with a β-blocker or calcium channel blocker, approximately 20% of patients with recent onset A-fib convert to
sinus rhythm. Because drugs that block normal AV conduction can accelerate conduction over the bypass tract, calcium channel or β-blocking drugs or digitalis are recommended only if there is reasonable certainty that a nodal bypass tract does not exist. Amiodarone is perhaps the best initial therapy for both rate control and rhythm conversion if a nodal bypass tract is suspected.
sinus rhythm. Because drugs that block normal AV conduction can accelerate conduction over the bypass tract, calcium channel or β-blocking drugs or digitalis are recommended only if there is reasonable certainty that a nodal bypass tract does not exist. Amiodarone is perhaps the best initial therapy for both rate control and rhythm conversion if a nodal bypass tract is suspected.
If A-fib is of less than 48-h duration, the ventricular rate is controlled; and it is judged that there is a reasonable likelihood of sustaining sinus rhythm and there are two reasonable courses of action. One is to perform synchronized cardioversion using 100 to 200 WS shock. Cardioversion is highly effective initially but unfortunately, A-fib recurs in most patients unless pharmacologic inhibition is continued; therefore, it makes little sense to convert patients intolerant of suppressive medications. The alternative course of action is to use amiodarone, ibutilide, or procainamide to chemically convert A-fib to sinus rhythm. Of the available choices, amiodarone is highly effective but has several practical limitations: when given IV, approximately 25% of patients develop hypotension and chemical phlebitis is common. Amiodarone has significant β-blocking properties and increases plasma levels of digoxin; both of which can lead to significant bradycardia after rhythm conversion. In addition, amiodarone potentiates the effects of warfarin and routinely results in abnormal thyroid function tests. Finally, amiodarone can cause pulmonary toxicity, especially when used in high doses or for long periods of time. Procainamide is effective in approximately 40% of patients but is generally poorly tolerated long-term. Ibutilide’s use is limited because the drug is only available parenterally and it rarely has a proarrhythmic effect, especially in patients with QT prolongation.
If A-fib has been present for more than 48 h, anticoagulation should be undertaken for 3 to 4 weeks before rhythm conversion so as to minimize the risk of embolization (unless atrial clot can be excluded with certainty using transesophageal echocardiography). After conversion, anticoagulation should be continued for another 3 to 4 weeks. For the patient in whom A-fib cannot be corrected, long-term anticoagulation is indicated to prevent systemic embolism and stroke. The annual incidence of stroke averages 1% for patients without mitral valve disease or heart failure but can be as high as 6% for patients with both risk factors.
Atrial Flutter
Atrial flutter (flutter) arises in a localized region of reentry outside the AV node or, less commonly, in a rapidly firing ectopic focus. Depolarization usually originates from low in the right atrium, producing inverted P waves in the inferior leads and upright P deflections in lead V1. Flutter frequently complicates pneumonia, exacerbations of chronic lung disease, and the postoperative course of thoracic surgery patients but seldom occurs following myocardial infarction (MI). Flutter is intrinsically unstable, often converting to A-fib spontaneously or in response to drug therapy. Because the flutter circuit does not involve the AV node, atrial rates are usually quite rapid (260 to 340 beats/min). The AV node cannot conduct impulses at such high rates, so the ventricular response is a fraction, typically 1/2 or 1/4 of the atrial rate. Most commonly, 2:1 AV block leads to a regular ventricular rate of approximately 150 beats/min. The ventricular response can be slowed, but the rhythm is rarely terminated by vagal maneuvers. If there is uncertainty about the rhythm, administration of adenosine is almost always diagnostic, revealing the characteristic “sawtooth” atrial depolarizations. Examination of the jugular pulse or recording of a right atrial pressure tracing can sometimes reveal the diagnostic atrial “flutter” waves.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


