Anesthesia for Neurosurgery
Gaganpreet Grewal
Michele Szabo
I. PHYSIOLOGY
A. Cerebral blood flow (CBF) is equal to cerebral perfusion pressure (CPP) divided by the cerebral vascular resistance. CPP is defined as the difference between the mean arterial pressure (MAP) and intracranial pressure (ICP) or central venous pressure, whichever is higher. CBF averages 50 mL/100 g of brain tissue per minute in the normal brain and is affected by blood pressure, metabolic demands, PaCO2, PaO2, blood viscosity, vasoactive agents, and neurogenic regulation. The brain receives approximately 15% of the cardiac output.
1. CBF is maintained at a constant level by constriction and dilation of arterioles (autoregulation) (Fig. 25.1) when the MAP is between 50 and 150 mm Hg. When MAP is outside these limits, CBF varies directly with the MAP. Chronic hypertension shifts the autoregulatory curve to the right, rendering hypertensive patients susceptible to cerebral ischemia at blood pressures considered normal in healthy individuals. Chronic antihypertensive therapy may normalize the autoregulatory range. Cerebral ischemia, trauma, hypoxia, hypercarbia, edema, mass effect, and volatile anesthetics attenuate or abolish autoregulation and may make blood flow to the affected area dependent on MAP.
2. PaCO2 has profound effects on CBF by its effect on the pH of the brain extracellular fluid (ECF). CBF increases linearly with increasing PaCO2 in the range from 20 to 80 mm Hg, with an absolute change of 1 to 2 mL/100 g/min for each mm Hg change in PaCO2. The effect of PaCO2 on CBF decreases over 6 to 24 hours because of slow adaptive changes in the brain ECF bicarbonate concentration. Sustained hyperventilation causes cerebrospinal fluid (CSF) bicarbonate production to decrease, allowing CSF pH to gradually normalize. Rapid normalization of PaCO2 after a period of hyperventilation results in a significant CSF acidosis with vasodilation and increased ICP.
3. PaO2. Hypoxia is a potent cerebral vasodilator. CBF increases markedly below a PaO2 of 60 mm Hg. PaO2 above 60 mm Hg has little influence on CBF.
4. Neurogenic regulation. The cerebral vasculature receives extensive cholinergic, adrenergic, serotonergic, and VIPergic innervation, although the exact role these systems play in regulation of CBF is not clear. Evidence suggests, however, that increased sympathetic tone in hemorrhagic shock shifts the lower end of the autoregulatory curve to the right and results in lower CBF at a given MAP.
5. Viscosity. Normal hematocrit (33% to 45%) in a normal brain has little influence on CBF. During focal cerebral ischemia, however, reduction in viscosity by hemodilution (hematocrit 30% to 34%) may increase CBF to ischemic territories.
B. Cerebral metabolic rate (CMRO2) and CBF are tightly coupled, because the brain requires a constant supply of substrate to meet its high metabolic demands. Regional or global increases in CMRO2 elicit a corresponding increase in CBF, probably mediated by signaling molecules such as
nitric oxide. Other factors that modulate CMRO2 (and CBF through this mechanism) include the following:
nitric oxide. Other factors that modulate CMRO2 (and CBF through this mechanism) include the following:
 FIGURE 25.1 Autoregulation maintains a constant level of CBF over a wide range of carotid artery mean blood pressures. Independent of this effect, CBF is elevated by hypercarbia (PaCO2) and hypoxemia (PaO2); hypocarbia diminishes CBF. |
1. Anesthetics. Variable effect (see sections II.A and B).
2. Temperature. Hypothermia decreases CMRO2 by 7% per 1°C below 37°C. Hyperthermia increases CMRO2.
3. Seizures. Increases CMRO2.
4. Pain or arousal. Increases CMRO2.
C. ICP reflects the relationship between the volume of the intracranial contents (brain, blood, and CSF) and the volume of the cranial vault. Normal ICP is 5 to 15 mm Hg. A sustained elevation of ICP greater than 15 to 20 mm Hg in the setting of intracranial pathology is considered abnormal.
1. The cranial vault is rigid, and its capacity to accommodate increases in intracranial volume is limited. A developing intracranial mass (e.g., tumor, edema, hematoma, or hydrocephalus) initially displaces blood and CSF, and ICP remains relatively normal (Fig. 25.2). As intracranial volume increases further, intracranial compliance decreases, and ICP rises rapidly (see Fig. 25.2). Thus, patients with decreased compliance may develop marked increases in ICP even with small increases in intracranial volume (e.g., cerebral vasodilation due to anesthesia, hypertension, or carbon dioxide retention) (see Fig. 25.2).
2. Clinical features of elevated ICP. ICP elevation usually decreases CPP and may cause ischemia in regions of the brain where autoregulation is defective and CBF depends on CPP. Early signs and symptoms of increased ICP include headache, nausea, vomiting, blurred vision, papilledema, and decreased levels of consciousness. As ICP increases, brain herniation may occur causing mechanical injury and/or ischemia to the brainstem and cranial nerves. This may result in hypertension with bradydysrhythmia or tachydysrhythmia, irregular respiration, oculomotor (third cranial) nerve palsy leading to ipsilateral pupillary dilation with no light reflex, abducens (sixth cranial) nerve palsy, contralateral hemiparesis or hemiplegia, and ultimately, coma and respiratory arrest.
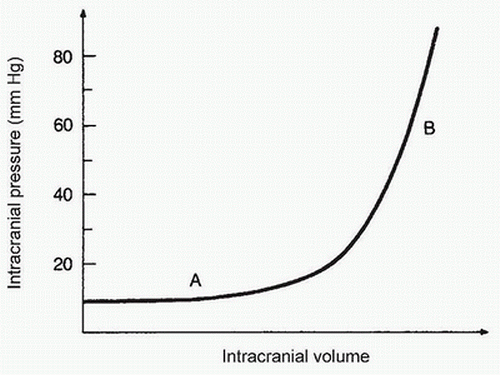 FIGURE 25.2 The intracranial compliance curve. In the normal ICP range (A), increases in intracranial volume produce minimal changes in ICP. Further, small increases in ICP after the “elbow” of the curve can produce abrupt increases in ICP (B). |
3. Treatment of elevated ICP involves strategies aimed at decreasing the volume of the intracranial components:
a. Hypoxia and hypercarbia cause cerebral vasodilation and should be avoided. Hyperventilation to a PaCO2 of 25 to 30 mm Hg produces cerebral vasoconstriction and may be used as a temporizing measure in the management of acutely increased ICP. Hyperventilation is potentially deleterious, however, and can cause ischemia in injured brain where CBF is low. Therefore, it should be withdrawn when effective definite therapy is established. Additionally, the effect of hyperventilation on ICP decreases as CSF pH normalizes.
b. Decrease jugular venous pressure. Elevating the head at least 30 degrees promotes venous drainage and decreases intracranial venous blood volume. Avoid excessive flexion or rotation of the neck and prevent increases in the intrathoracic pressure (e.g., coughing, straining, and elevated intrathoracic pressure). Positive end-expiratory pressure should be minimized to the lowest level that maintains adequate lung recruitment.
c. Control CMRO2. Barbiturates are potent vasoconstrictors that decrease cerebral blood volume while decreasing CMRO2. Prevent increases in CMOR2 due to arousal/seizures with adequate sedation and seizure prophylaxis where indicated.
d. Maintaining high serum osmolality (305 to 320 mOsm/kg) may reduce cerebral edema and decrease brain volume. Fluid management is designed to achieve this goal (see section V.D). In addition, mannitol (0.5 to 2.0 g/kg intravenous [IV]) and furosemide produce a hyperosmolar state and are effective in the acute reduction of ICP. Hypertonic saline is an alternative to mannitol for managing raised ICP.
e. CSF volume can be reduced by draining the CSF through ventriculostomy catheter or needle aspiration intraoperatively.
f. Surgical removal of tumor, hematoma, or decompressive craniectomy reduces intracranial volume and ICP.
g. Steroids may reduce cerebral edema associated with tumors.
II. PHARMACOLOGY
Agents used in anesthesia may affect CMRO2 and CBF.
A. Inhalation anesthetics produce a dose-related reduction in CMRO2 while causing an increase in CBF.
1. Nitrous oxide can cause increases in CMRO2, CBF, and ICP. These effects can be greatly attenuated or abolished when it is administered in conjunction with IV anesthetic agents. Nitrous oxide should be avoided when intracranial airspaces (e.g., pneumocephalus) exist, because it diffuses more rapidly into such spaces than nitrogen diffuses out and may produce an acute increase in ICP.
2. Volatile agents cause increases in CBF due to their direct vasodilatory actions. Autoregulation can be attenuated or abolished by increasing the concentrations of these drugs, but responsiveness to carbon dioxide seems to be preserved (Table 25.1). The vasodilatory effect of inhalational agents is clinically insignificant in patients with normal intracranial compliance. These agents should be used with caution in patients with compromised intracranial compliance (e.g., large intracranial mass lesion and acute intracranial hematoma).
3. Volatile anesthetics produce dose-dependent reductions in metabolism (CMRO2), probably by depressing neuronal electrical activity. Isoflurane is the most potent in this respect and is the only volatile agent that induces an isoelectric electroencephalogram (EEG) at clinically relevant concentrations (2 × MAC).
B. IV anesthetics generally cause coupled reduction in CBF and CMRO2 in a dose-dependent manner. This is due to the depression of cerebral metabolism. Barbiturates, etomidate, and propofol markedly decrease CBF and CMRO2 and can produce isoelectric EEGs. Etomidate has been associated with seizures and is best avoided in seizure-prone patients. Lidocaine in therapeutic doses decreases both CBF and CMRO2. Ketamine, by contrast, increases CBF and CMRO2 and is used infrequently in neuroanesthesia. Opioids and benzodiazepines produce minimal changes in CBF and CMRO2. Autoregulation and carbon dioxide responsiveness appear to be preserved with IV agents.
C. Muscle relaxants have no direct effect on CBF and CMRO2. They may alter cerebral hemodynamics indirectly through their effects on blood pressure. Succinylcholine produces a transient increase in ICP, likely caused by arousal phenomena, which can be attenuated by prior administration of a barbiturate or a defasciculating dose of a nondepolarizing muscle relaxant.
D. Vasoactive Drugs
1. Adrenergic agonists. α-Adrenergic agonists and low-dose β-adrenergic agonists have little influence on CBF if MAP is within the limits of
autoregulation. Larger doses of β-adrenergic agonists can produce an increase in CMRO2 and CBF that can be exaggerated in the setting of a defect in the blood-brain barrier. Dopamine causes an increase in CBF with little change in CMRO2.
autoregulation. Larger doses of β-adrenergic agonists can produce an increase in CMRO2 and CBF that can be exaggerated in the setting of a defect in the blood-brain barrier. Dopamine causes an increase in CBF with little change in CMRO2.
TABLE 25.1 Cerebral Physiologic Effects of Inhalational Anesthetics | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
2. Vasodilators. Sodium nitroprusside, nitroglycerin, hydralazine, nimodipine, and nicardipine can increase CBF and ICP by direct cerebral vasodilation if MAP is maintained. β-Adrenergic-blocking agents probably have minimal effects. Despite these profiles, all these agents have been used safely during neuroanesthesia, particularly if CPP is maintained.
E. Cerebral Protection
1. Focal versus global cerebral ischemia
a. Focal ischemia is characterized by an area of densely ischemic tissue that is surrounded by nonischemic brain, which may provide collateral flow to the penumbral margins. This residual blood flow may allow neurons to survive for varied periods of time (e.g., thrombolysis within 3 hours after stroke onset may prevent a full infarct due to reperfusion).
b. Complete global ischemia is characterized by absent CBF (e.g., cardiac arrest). Tolerance for surviving global ischemia is on the order of minutes.
2. Agents
a. IV anesthetic agents: High-dose barbiturates may slightly improve neurologic recovery from focal ischemia, possibly by decreasing metabolic rate or more likely by a direct pharmacologic effect. Propofol may also reduce focal ischemic cerebral injury, although it is not as extensively studied as the barbiturates. Etomidate aggravates ischemic brain injury. Early clinical reports suggest that prophylactic low-dose lidocaine may have neuroprotective effects in nondiabetic patients.
b. Volatile anesthetic agents may provide some cerebral protection, but data are conflicting, and it is unclear whether this neuroprotection is sustained.
c. Nimodipine’s beneficial effects on vasospasm after subarachnoid hemorrhage (SAH) are well established and are likely mediated through neuronal, rather than vascular, effects. Clinical trials failed to detect a beneficial effect for acute stroke patients.
d. Steroids have not been found to be beneficial after stroke or severe head injury.
e. Magnesium confers significant neuroprotection in animal studies. However, a large clinical trial did not show protection in acute stroke victims.
f. Hypothermia reduces metabolism for both neuronal and cellular functions and therefore may be beneficial in the setting of decreased cerebral perfusion. Induced mild hypothermia (12 to 24 hours) has been shown to be effective in reducing morbidity in patients who sustain cardiac arrest. By contrast, two clinical studies did not demonstrate improved outcome when induced mild hypothermia was used in patients after significant head injury or intraoperatively for aneurysm surgery.
g. Hyperthermia profoundly worsens outcome from focal cerebral ischemia and should be avoided.
h. Moderate hyperglycemia (>170 mg/dL) exacerbates neurologic injury after an ischemic insult. There are human data suggesting that normalizing blood glucose causes higher incidence of good outcome in stroke patients.
i. Other physiologic variables: In addition to the above-mentioned variables of temperature and glucose, meticulous management of perfusion pressure, pCO2, pO2, pH normalization, and seizure prophylaxis contribute significantly to improved neurologic outcome in the setting of cerebral ischemia. Maintenance of a high-normal CPP can augment collateral CBF. By contrast, hypotension reduces CBF and exacerbates the injury. Normocapnia should be maintained. Seizures, which can increase CBF and ICP and decrease CPP, should be prevented and rapidly treated.
3. There is preliminary evidence that female sex hormones may confer some neuroprotection after TBI.
III. ELECTROPHYSIOLOGIC MONITORING
A. Electroencephalography (EEG) measures electrical activity of the neurons of the cerebral cortex and is thus used as a threshold marker for detecting ischemia due to inadequate CBF. It is used frequently during procedures that jeopardize cerebral perfusion, such as carotid endarterectomy, or to ensure electrical silence before circulatory arrest.
1. Normal CBF in gray and white matter averages 50 mL/100 g/min. With most anesthetic techniques, the EEG starts to become abnormal when the CBF decreases to 20 mL/100 g/min. Isoflurane is distinct as the EEG becomes abnormal when CBF is much lower at 8 to 10 mL/100 g/min. Cellular survival is endangered when CBF decreases to 12 mL/100 g/min (lower with isoflurane). Thus, EEG changes can warn of ischemia before CBF becomes insufficient to maintain tissue viability. Prompt detection of EEG changes may be treated with increases in perfusion pressure or shunting to restore CBF to prevent infarction.
2. The EEG may exhibit changes intraoperatively with no demonstrable neurologic deficit during postoperative examination. Cerebral ischemia can produce electrical dysfunction without causing neuronal cell damage because the blood flow threshold for electrical failure is higher than that needed to maintain cellular integrity.
3. Factors other than anesthetics that may affect the EEG include hypothermia (which may limit the usefulness of EEG during cardiopulmonary bypass), hypotension, hypoglycemia, hypoxia, tumors, vascular abnormalities, and epilepsy. An abnormal EEG in patients with preexisting neurologic deficits, strokes in evolution, and recent reversible ischemic neurologic deficits can also make it difficult to interpret new changes.
4. Anesthetic effects on the EEG are generally global, which often helps distinguish them from the focal changes of ischemia. A predominance of slow activity is seen as the anesthetic depth increases. “Deep” anesthesia may cause marked EEG slowing, making detection of superimposed ischemic changes during critical periods difficult to interpret. Maintaining a constant level of anesthesia during critical periods (e.g., carotid clamping) facilitates EEG interpretation.
B. Evoked Potential Monitoring
1. Sensory-evoked potentials (SEPs) are electrical potentials generated within the neuraxis in response to stimulation of a peripheral or cranial nerve. As they travel from the periphery to the brain, these potentials can be recorded by electrodes placed over the scalp and along the transmission pathway. Evoked potentials (EPs) have lower voltage than background EEG activity, but summation of hundreds of signals using computerized devices makes it possible to extract them by averaging
out the random background EEG. A normal response implies that the conduction pathway is intact. Damage to the pathway generally decreases the amplitude or prolongs latency (i.e., the time from peripheral stimulus to arrival of potentials at the recording site) of the waveform peaks. EPs are classified according to the nerve tract being evaluated.
out the random background EEG. A normal response implies that the conduction pathway is intact. Damage to the pathway generally decreases the amplitude or prolongs latency (i.e., the time from peripheral stimulus to arrival of potentials at the recording site) of the waveform peaks. EPs are classified according to the nerve tract being evaluated.
a. Somatosensory-evoked potentials (SSEPs) are obtained by stimulating a peripheral nerve (e.g., median nerve at the wrist or posterior tibial nerve at the ankle or in the popliteal fossa) and recording the elicited signals over the spinal cord (spinal SSEPs) or cerebral cortex (cortical SSEPs). SSEPs are used most commonly to monitor spinal cord function during spinal cord or vertebral column surgery (e.g., major spine surgery with instrumentation) and may be used during peripheral nerve, brachial plexus, or thoracic aortic surgery (to detect spinal ischemia during aortic cross-clamping). Because SSEPs are conducted primarily by the dorsal column in the spinal cord, there are concerns about the reliability of SSEP monitoring for detecting threatened motor function (i.e., anterior spinal cord ischemia). For this reason, the “wake-up test” is used in some centers (see section VII.B.2), as well as motor-evoked potential (MEP) monitoring.
b. Brainstem-auditory evoked potentials (BAEPs) are recorded by delivering an auditory stimulus to one ear through an ear-insert headphone. BAEPs reflect the transmission of electrical impulses along the auditory pathway and are monitored during posterior fossa surgery in an attempt to avoid brainstem or auditory (eighth cranial) nerve damage.
2. MEPs. Monitoring the integrity of the motor tracts within the spinal cord may be more reliable than SSEP monitoring during spinal surgery. The ventral motor columns of the spinal cord may be more susceptible to ischemia than the posterior proprioceptive fibers. Motor impulses can be generated by transcranial electrical stimulation. The evoked responses are measured as a potential over the spinal cord below the surgical field and in the muscle of interest. Anesthetics substantially modify transcranially induced potentials but less so if the stimulus is measured in the spinal cord below the surgical field.
3. Electromyography (EMG) records muscle responses to the stimulation of motor nerves. EMG is used frequently when there is risk of facial nerve injury during cerebellopontine angle surgery (e.g., posterior fossa surgery for meningioma). Because the EMG records motor responses to stimulation, neuromuscular-blocking agents are avoided during the periods of electrical stimulation.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


