Hormones Produced by the Adrenal Cortex
Three main classes of hormones are produced by the adrenal cortex:
glucocorticoids (e.g., cortisol),
mineralocorticoids (e.g., aldosterone), and
sex steroids (e.g., testosterone,
DHEA, and androstenedione).
All steroid hormones are derived from the cyclopentanoperhydrophenanthrene structure that is composed of three cyclohexane rings and a single cyclopentane ring. Although adrenal steroid cells can synthesize cholesterol de novo from acetate,
80% of the cholesterol precursor for adrenal cortex hormone synthesis is provided from circulating plasma lipoproteins. Cholesterol (mainly LDL in circulation) is the precursor for all adrenal steroidogenesis (
14).
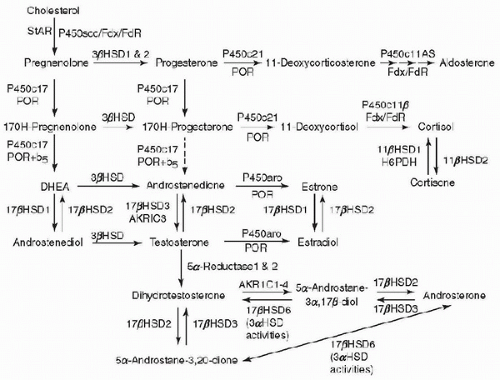
The biochemical pathways involved in adrenal steroidogenesis are shown in
Figure 105.1, and the adrenal steroidogenic enzymes and cofactors are illustrated in
Table 105.1. Conversion of cholesterol to pregnenolone has been described as the rate-limiting step for steroidogenesis, catalyzed by side-chain cleavage enzyme.
However, the true rate-limiting step of adrenal steroidogenesis is the transfer of cholesterol across the inner mitochondrial membrane. This requires the action of the steroidogenic acute regulatory (StAR) protein. At the mitochondrial inner membrane, the side chain of cholesterol is then cleaved to yield pregnenolone, catalyzed by cholesterol side-chain cleavage enzyme (cholesterol desmolase, P450scc, CYP11A1), a cytochrome P450 (CYP) enzyme (
15). Pregnenolone then diffuses out of mitochondria and enters the endoplasmic reticulum. The subsequent reactions that occur are zone dependent and are mediated by two groups of enzymes and cofactors (cytochrome P450 enzymes and hydroxysteroid dehydrogenase). These enzymes undergo posttranslational modification with the assistance of electron-donating cofactors. There are two types of P450 enzymes; type 1 P450 enzymes that receive electrons from NADPH via ferredoxin and ferredoxin reductase and type 2 P450 enzymes that receive electron from P450 oxidoreductase (
POR) (
16). In the
zona glomerulosa, pregnenolone is converted to progesterone by 3
β-hydroxysteroid dehydrogenase (HSD3B2). Progesterone is converted to 11-deoxycorticosterone by steroid 21-hydroxylase (P450c21, CYP21), which is another cytochrome P450 enzyme. In the mitochondria, deoxycorticosterone is then converted to aldosterone by aldosterone synthase (P450aldo, CYP11B2). Aldosterone synthase also performs three successive oxidations: 11
β-hydroxylation, 18-hydroxylation, and further oxidation of the 18-methyl carbon to an aldehyde. In the
zona fasciculata, pregnenolone and progesterone are converted by 17
α-hydroxylase (P450c17, CYP17) to 17-hydroxypregenolone and 17-hydroxyprogesterone, respectively, in the endoplasmic reticulum. This enzyme is not expressed in the zona glomerulosa. 17-Hydroxypregnenolone is converted to 17-hydroxyprogesterone and 11-deoxycortisol by the same 3
β-hydroxysteroid and 21-hydroxylase enzymes, respectively, which are active in the zona glomerulosa. 11-Deoxycortisol is converted to cortisol in the mitochondria by steroid 11
β-hydroxylase (P450c11, CYP11B1). In the
zona reticularis and to some extent in the
zona fasciculata, the 17-hydroxylase (CYP17) enzyme has an additional activity, which is the cleavage of the 17,20-carbon-carbon bond. 17-Hydroxypregnenolone is converted by 3
β-hydroxysteroid dehydrogenase to androstenedione.
Regulation of the Adrenal Cortex (Hypothalamic-Pituitary-Adrenal Axis)
The major regulator of glucocorticoid secretion is by
ACTH.
ACTH is a 39-amino acid peptide that is produced in the anterior pituitary (
17), and is synthesized as a part of a largemolecular-weight precursor proopiomelanocortin (
POMC).
ACTH is released in bursts, which vary in amplitude throughout the 24-hour cycle. The normal diurnal rhythm of cortisol secretion is established after infancy. In children and adults, the pulses of
ACTH and cortisol are the highest in the early morning hours, become lower in late afternoon, and in the evening reach their nadir 1 or 2 hours after sleep begins.
ACTH secretion from the anterior pituitary is stimulated mainly by
CRH. This hormone is synthesized by neurons of the parvocellular division of the hypothalamic paraventricular nucleus (
17,
18). The secretion of
ACTH and
CRH is predominantly regulated by cortisol through a negative feedback effect.
ACTH can also inhibit its own secretion.
Aldosterone secretion is regulated mainly by the renin-angiotensin system and by serum potassium levels (
19,
20).
ACTH plays a very small role in the regulation of its synthesis. Predominantly, in response to decreased intravascular volume, as in dehydration, renin is secreted by the juxtaglomerular apparatus of the kidney. Renin is a proteolytic enzyme that cleaves an
α2-globulin produced by the liver called angiotensinogen and results in the formation of angiotensin I, which is cleaved further by angiotensin-converting enzyme (
ACE) in the lungs and other tissues yielding the biologically active angiotensin II. Angiotensin II is cleaved further to produce the angiotensin III. Angiotensin II and III are potent stimulators of aldosterone secretion. Angiotensin II occupies a G protein-coupled receptor and activates phospholipase C. Phospholipase C triggers a cascade, which results in a rise in the intracellular calcium and activates protein kinase C and calmodulin-activated (CaM) kinases. CaM kinases increase transcription of aldosterone synthase (CYP11B2), the enzyme needed for aldosterone synthesis (
21).
Adrenal Steroid Action
Both cortisol and aldosterone act by binding to intracellular receptors: the glucocorticoid receptor and the mineralocorticoid receptor. These receptors belong to the superfamily of nuclear receptors. They have a common structure that contains a C-terminal ligand-binding domain, a central DNA-binding domain, and an N-terminal hypervariable region. The binding of steroid to its receptor in the cytoplasm results in its dimerization and translocation to the nucleus. In the nucleus, they bind the glucocorticoid response element on the glucocorticoid responsive genes, which results in increased transcription.
Glucocorticoids have regulating effects on carbohydrate, lipid, and protein metabolism. They increase hepatic gluconeogenesis, glycolysis, proteolysis, and lipolysis. Glucocorticoids can result in increased insulin levels, which will inhibit peripheral tissue glucose uptake leading to hyperglycemia. In addition, glucocorticoids may work in parallel to insulin by stimulating glycogen deposition and production in the liver, which provides protection against starvation. An increase in free fatty acid levels is associated with glucocorticoid administration. This results from glucocorticoid enhancement of lipolysis, decrease of cellular glucose uptake, and decrease in glycerol production. In addition, there is an increase of amino acid substrates that are used in gluconeogenesis due to proteolysis in fat, skeletal muscle, bone, lymphoid, and connective tissues.
Glucocorticoid excess can decrease the levels of free IGF-1 and increase IGFBP-1 resulting in a decrease in free IGF-1. They also exert a direct inhibitory effect on the epiphyses, which results in delayed skeletal maturation and decreased linear growth in children. Although excess glucocorticoids can impair growth, they are also essential for normal growth and development. In the fetus and neonate, they accelerate the differentiation and development of various tissues, such as the development of the hepatic and gastrointestinal systems, as well as the production of surfactant in the fetal lung.
Glucocorticoids also play a major role in immune regulation. They suppress the inflammatory process. Depletion of monocytes, eosinophils, and lymphocytes (T lymphocytes) is observed with the administration of high doses of glucocorticoids. T lymphocytes are reduced more than the B lymphocytes, leading to a predominantly humoral immune response. Glucocorticoids also inhibit immunoglobulin synthesis and stimulation of lymphocyte apoptosis (
22). In addition, they block other anti-inflammatory effects, such as histamine and proinflammatory cytokine secretion (e.g., tumor necrosis factor-
α, interleukin-1, and interleukin-6).
Glucocorticoids have a positive inotropic effect on the heart that leads to an increase in left ventricular output. They also increase blood pressure by a number of mechanisms that involve the vascular system and the kidneys. In the vascular smooth muscles and the heart, glucocorticoids have a permissive effect on the actions of epinephrine and norepinephrine. They also increase the sensitivity to vasopressor agents, such as catecholamines and angiotensin II, while reducing nitric oxide-mediated endothelial dilatation (
23). Hypertension is often observed in patients with glucocorticoid excess; it is thought to be due to the activation of mineralocorticoid receptor.
Glucocorticoids can induce a negative calcium balance by increasing renal calcium excretion and inhibition of calcium absorption by the intestine. Long-term use of glucocorticoid can lead to osteopenia and osteoporosis as they also inhibit the osteoblastic activity. Glucocorticoids also have effects on brain metabolism and mood changes such as emotional liability with irritability, euphoria as well as appetite stimulation and insomnia can occur.
The major role of mineralocorticoids is to maintain intravascular volume. This is achieved by sodium retention coupled with the elimination of potassium and hydrogen ions. The main target tissues for the action of mineralocorticoids are the kidney, gut, and salivary and sweat glands. Mineralocorticoids act mainly on the distal convoluted tubules and cortical collecting ducts of the kidney. They stimulate the reabsorption of sodium and the secretion of potassium in the distal convoluted tubules. The mineralocorticoid receptor has a similar affinity for cortisol and aldosterone, yet glucocorticoids have limited mineralocorticoid activity. This is due to the action of 11β-hydroxysteroid dehydrogenase type 2, which converts cortisol to cortisone. Cortisone does not, under normal circumstances, occupy the mineralocorticoid receptor.
 Biology of the adrenal function
Biology of the adrenal function Adrenocortical insufficiency
Adrenocortical insufficiency Relative adrenal insufficiency
Relative adrenal insufficiency Adrenal hyperfunction
Adrenal hyperfunction Steroid supplementation during critical illness
Steroid supplementation during critical illness Therapeutic guidelines and steroid preparations
Therapeutic guidelines and steroid preparations