Adrenal Disorders
Anzar Haider
Adrenal disorders represent a classical bio-feedback system and underscore the importance of physiology and biochemistry in understanding the disease process.
ANATOMY
The adrenal gland is a pyramidal-shaped organ that lies above the upper pole of the kidneys. It functionally consists of two endocrine tissues; the outer one derived from mesodermal tissue, develops into the adrenal cortex; and the inner one derived from ectodermal neural crests forms the medulla.
The fetal adrenal cortex forms by the fifth week of gestation, consisting of two distinct zones, an outer zone “adult/definite zone” that produces glucocorticoids and mineralocorticoids, and a much larger “fetal zone” that mainly produces androgens (dehydroepiandrosterone [DHEA]) for the synthesis of estrogen by the placenta. The fetal zone regresses after birth and the adult/definite zone then proliferates and androgen-specific reticularis zone develops by 5 to 6 years of age.
PHYSIOLOGY AND REGULATION OF THE ADRENAL CORTEX
The adrenal cortex in older children and adults is divided into three zones:
Zona glomerulosa: Outer zone that principally produces aldosterone
Zona fasciculata: Middle zone that synthesizes glucocorticoids
Zona reticularis: Inner zone that principally secretes androgens (DHEA and androstenedione)
All adrenal hormones are synthesized from cholesterol by a series of P450 (CYP) and hydroxysteroid dehydrogenase (HSD) enzymes. Aldosterone is regulated by the renin-angiotensin system and is influenced by sodium levels and blood pressure. Cortisol secretion is under the control of adrenocorticotrophic hormone (ACTH), which in turn is stimulated by corticotropin releasing hormone (CRH) from the hypothalamus. No direct stimulatory signal accounts for the release of adrenal androgen secretion; however, ACTH does have a permissive role in the secretion of adrenal androgens.
DISORDERS OF ADRENAL CORTEX
Adrenal cortical secretions consist of glucocorticoid, mineralocorticoid, and androgen and can be broadly classified into hypofunctional and hyperfunctional states.
Adrenal insufficiency (hypofunction) is termed primary when the defect is in the adrenal gland itself, and secondary if the defect is in the pituitary region.
Primary Adrenal Insufficiency (Low Cortisol, High Adrenocorticotrophic Hormone)
Patients with primary adrenal insufficiency may present in adrenal crises with cardiovascular collapse and can be fatal. Clinical features are the result of combined defects in glucocorticoid and mineralocorticoid secretion. There are congenital and acquired causes.
Congenital Primary Adrenal Insufficiency
Causes of primary adrenal insufficiency include:
Congenital adrenal hypoplasia
Congenital adrenal hyperplasia (CAH)
Familial glucocorticoid deficiency (ACTH resistance)
Metabolic disease: Adrenoleukodystrophy, Wolman disease, Smith Lemli Opitz
Infants with congenital adrenal hypoplasia present in the newborn period, typically 2 weeks after birth, with hypoglycemia, jaundice, failure to thrive, and vomiting. These patients have severe glucocorticoid, mineralocorticoid, and androgen deficiency. Thus, they may present with dehydration, hypotension, hyponatremia, and hyperkalemia.
Males are generally affected, as this is caused by an Xlinked mutation in the DAX-1 gene on chromosome Xp21. This gene is important in the development of the adrenal glands and gonads. The gene is contiguous to the glycerol kinase and Duchenne muscular dystrophy gene. Recently, an autosomal gene (SF-1) also has been identified; heterozygous mutation causes adrenal and gonadal hypoplasia.
Males are generally affected, as this is caused by an Xlinked mutation in the DAX-1 gene on chromosome Xp21. This gene is important in the development of the adrenal glands and gonads. The gene is contiguous to the glycerol kinase and Duchenne muscular dystrophy gene. Recently, an autosomal gene (SF-1) also has been identified; heterozygous mutation causes adrenal and gonadal hypoplasia.
Congenital adrenal hyperplasia is a group of inherited autosomal recessive disorders of one of the enzyme in the cortisol pathway that results in hyperplasia of the adrenal gland with low cortisol and an elevated ACTH level. Adrenal steroidogenesis pathway is depicted in (Fig. 16.1). Enzyme defects are discussed later in detail.
Familial glucocorticoid deficiency is transmitted in autosomal recessive manner. Patients have ACTH resistance and may have mutations in the MC2 receptor.
Adrenoleukodystrophy (ALD) is an X-linked disorder characterized by progressive CNS demyelination and adrenal failure. Signs of adrenal failure may be the initial and only manifestation of this disorder, although neurologic symptoms may precede the adrenal deficiency. It is caused by a gene mutation on chromosome Xq28. The diagnosis is established by finding an elevated level of very long chain fatty acids (VLCFA) in the plasma. This condition should be ruled out in all males presenting with adrenal insufficiency.
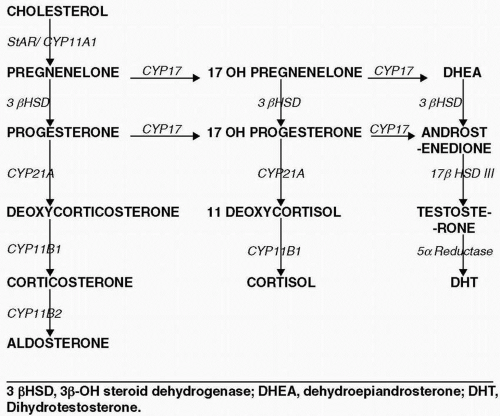 Figure 16.1 The adrenal steroidogenesis pathway. |
Triple A syndrome (Allgrove syndrome) consists of ACTH resistance, achalasia, and alacrima. Many patients may have neurologic symptoms as well. This syndrome is caused by mutation in the AAAS gene whose product ALADIN is expressed in the central nervous (CNS) and gastrointestinal systems.
Acquired Primary Adrenal Insufficiency
Causes of acquired primary adrenal insufficiency include:
Autoimmune adrenalitis (isolated)
Autoimmune polyglandular syndrome (APS) Type 1 and Type 2
Adrenal infections (TB, HIV, fungal, sepsis)
Adrenal hemorrhage, infarction, and infiltration
Medications (mitotane, ketoconazole, megestrol, medroxyprogesterone)
Autoimmune adrenalitis is the most common cause of acquired adrenal insufficiency. It may occur as an isolated problem or as a part of the autoimmune polyglandular syndrome (APS). There are two types associated with adrenal insufficiency:
Type I APS typically presents in younger patients (first decade of life) and is also called APECED (autoimmune polyendocrinopathy with cutaneous ectodermal dysplasia). This is caused by a mutation in the AIRE gene on chromosome 21. It is characterized mainly by:
Hypoparathyroidism
Adrenal insufficiency
Mucocutaneous candidiasis
Other minor manifestations: primary hypogonadism, Hashimoto thyroiditis, vitiligo, alopecia
Type II APS is characterized by:
Hypothyroidism
Adrenal insufficiency
Type I diabetes mellitus
Other endocrine organs also may be affected. It occurs in late adolescence and appears to have an autosomal dominant pattern of transmission. Adrenal failure is typically the first manifestation.
Secondary Adrenal Insufficiency (Low Cortisol, Low Adrenocorticotrophic Hormone)
Signs and symptoms of secondary adrenal insufficiency are non-specific and milder than in primary adrenal insufficiency. These can also be classified into congenital and acquired.
Congenital causes include:
Hypopituitarism
Septo-optic dysplasia
Cerebral midline defects
Isolated ACTH defect
Acquired causes include:
Craniopharyngioma
Cranial irradiation
Infiltrative pituitary and hypothalamic lesions (Langerhans cell histiocytosis, sarcoidosis)
Hypophysitis
Surgery to the hypothalamic-pituitary area
Steroid therapy
Mineralocorticoid function in these disorders is intact; thus, these patients do not present with salt wasting and cardiovascular collapse. ACTH deficiency typically presents in the context of multitrophic hypothalamic pituitary disease. Neonates may present with hypoglycemia, poor feeding, and jaundice. Older children may have nonspecific symptoms of inappropriate fatigue, anorexia, weakness, ill-defined abdominal pain, nausea, and prolonged recovery from illness. Symptoms of orthostatic hypotension, salt craving, and generalized skin pigmentation— characteristics of primary adrenal insufficiency—are not found in secondary adrenal insufficiency.
Laboratory Investigations of Adrenal Insufficiency
Laboratory findings may include a low morning cortisol concentration, associated with an elevated ACTH concentration in primary, but a normal or low ACTH in secondary adrenal insufficiency. Mineralocorticoid insufficiency occurs in primary adrenal insufficiency and can be diagnosed by an increased plasma renin activity, low aldosterone, sodium, and bicarbonate levels, and high potassium level. Since mineralocorticoid activity is normal in secondary adrenal insufficiency, hyperkalemia is not present, but hyponatremia occurs because of decreased water excretion from a lack of glucocorticoid activity.
Treatment of Adrenal Insufficiency
Hydrocortisone 10 to 15mg/m2/day should be given two to three times a day to replace glucocorticoid deficiency. Fludrocortisone should be added in mineralocorticoid deficiency (typically found in primary adrenal insufficiency states).
Glucocorticoid Excess (Hypercortisolism)
Cushing syndrome is a disorder of excess glucocorticoid secretion and the effect of prolonged exposure to cortisol. Cushing syndrome is rare in children. The most common cause in children is exogenous administration of pharmacological dose of glucocorticoid.
Endogenous Cushing syndrome includes Cushing disease if the source is increased production from the pituitary caused by an ACTH-secreting pituitary microadenoma. This accounts for 75% of all cases of Cushing syndrome in children over 7 years of age. In children under 7 years of age, adrenal sources of Cushing syndrome are common (adrenal adenoma, carcinoma, or bilateral hyperplasia). Most adrenal tumors in children presenting with Cushing syndrome are malignant.
Clinical features include weight gain, growth deceleration, fatigue, muscle weakness, and emotional lability. The patient may have increased facial plethora, easy bruising, violaceous striae, hirsutism, and hypertension. Growth failure in Cushing syndrome is the most important distinguishing feature from exogenous obesity and can obviate the need for an expensive workup for obesity.
Elevated values of midnight salivary cortisol, 24-hour urinary free cortisol, and cortisol level drawn in the early morning following overnight dexamethasone administration (overnight dexamethasone test) can be used to screen for hypercortisolemia. The low-dose, high-dose
dexamethasone suppression test can be employed to distinguish pituitary versus adrenal source. Cortisol values are not suppressed in disorders of adrenal origin.
dexamethasone suppression test can be employed to distinguish pituitary versus adrenal source. Cortisol values are not suppressed in disorders of adrenal origin.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


