Acute Endocrine Complaints
Pavel Fort MD
Phyllis W. Speiser MD
PART 1 Adrenal Insufficiency and Crisis
Phyllis W. Speiser MD
INTRODUCTION
Adrenal insufficiency is important to recognize because of its potentially life-threatening implications. Symptoms and signs of this disorder are varied and nonspecific. In infancy, these include lethargy, vomiting, poor appetite, and failure to thrive. Clinicians may mistake these problems for formula intolerance or inadequate lactation, or alternatively, primary infectious or gastrointestinal disorders. In older children, chronic fatigue, headache, gastrointestinal symptoms, salt craving and hyperpigmentation may be noted. Patients may undergo extensive evaluation before a diagnosis is made.
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
The adrenal glands, located at the upper pole of each kidney with a combined weight of 10 g in the adult, are composed of cortex and medulla. The adrenal cortex is the primary site of cortisol and aldosterone synthesis and a secondary site of sex hormone synthesis. The adrenal medulla is responsible for catecholamine synthesis, epinephrine, and norepinephrine, important in nervous system regulation. This section is restricted to discussion of adrenal cortical disease.
Glucocorticoids, such as cortisol, are essential for survi-val, mediating myriad developmental and physiologic processes. Some actions of cortisol include stimulating gluconeogenesis and glycogenolysis. By increasing cellular breakdown of sugars, protein, and fat, more substrate is provided for gluconeogenesis. Glucocorticoids also increase cellular resistance to insulin, thereby decreasing glucose uptake and use in peripheral tissues. The overall effect is to raise and maintain blood glucose levels, especially during stress (Pilkis & Granner, 1992). Thus, glucocorticoid deficiency may cause hypoglycemia.
Glucocorticoids also increase cardiac output and enhance vascular sensitivity to pressor hormones (Kelly, Mangos, Williamson, & Whitworth, 1998). Hence, glucocorticoid deficiency reduces cardiac output and may predispose the patient to heart failure and shock.
These problems are exacerbated by concomitant aldosterone deficiency. Aldosterone is essential for normal sodium homeostasis; deficiency of this hormone results in sodium loss through the kidney, colon, and sweat glands (Bonvalet, 1998).
Cortisol and aldosterone are synthesized from cholesterol in the adrenal cortex in several enzymatic steps (see Figure 47-1). Aldosterone synthesis differs from cortisol synthesis in that a specific 11β-hydroxylase isozyme (enzyme), termed aldosterone synthase, is used at the terminal step. Aldosterone synthesis is separately regulated, principally by the renin-angiotensin system. Cortisol synthesis is regulated primarily by pituitary adrenocorticotropic hormone (ACTH) and hypothalamic corticotropin-releasing hormone (CRH). Insufficient production of these tropic hormones or unresponsiveness to their actions results in select adrenocortical hormone deficiencies. Enzymatic blocks impairing cortisol synthesis or resistance to cortisol’s actions also presents as adrenal insufficiency (Speiser & White, 1998). Excessive ACTH secretion is a consequence of cortisol deficiency, causing both adrenal hyperplasia and hyperpigmentation. If aldosterone synthesis is blocked or if there is selective resistance to aldosterone action, sodium wasting and failure to thrive will result without complete adrenocortical insufficiency.
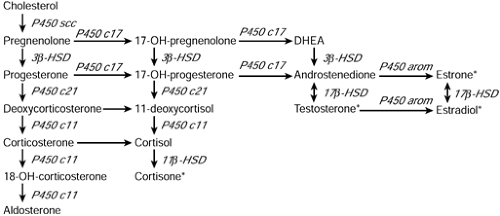 Figure 47-1 Diagram of the pathways of corticosteroid synthesis. Cholesterol is converted in several steps to aldosterone, cortisol, or sex steroids. Hormones marked by an asterisk (*) are produced largely outside the adrenal cortex. Deficiency of a given enzyme causes accumulation of hormonal precursors and a deficiency of products. DHEA = dehydroepiandrosterone; HSD = hydroxysteroid dehydrogenase. Adapted with permission from Speiser, P.W. (1995). Congenital adrenal hyperplasia. In K.L. Becker (ed.). Principles and practice of endocrinology and metabolism, 2nd ed. Philadelphia: J.B. Lippincott. |
Secondary Adrenal Insufficiency
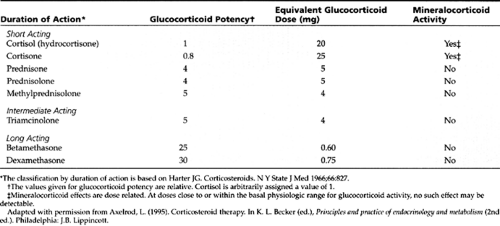
This refers to an externally induced alteration in pituitary structure or function preventing normal ACTH secretion and thus causing adrenal insufficiency. A frequent cause of iatrogenic secondary adrenal insufficiency, both acute and chronic, is withdrawal of exogenous glucocorticoid therapy. Table 47-1 shows the most commonly used glucocorticoids, their relative potencies, equivalent doses, and mineralocorticoid actions.
 |
Administration of hydrocortisone, prednisone, or dexamethasone in supraphysiologic doses (eg, prednisone = 10 mg daily) for periods of greater than 2 to 3 weeks requires gradual tapering to allow the hypothalamic-pituitary-adrenal (HPA) axis time to recover from suppression.
• Clinical Pearl
Short courses of steroids (eg, asthma treatment with oral steroids over 5–7 days) do not require long-term tapering (Orth & Kovacs, 1998).
Steroid tapering should be individualized because each patient tolerates reduction of treatment differently; this will depend on the total dosage, dose schedule, and duration of steroid treatment. Usually, a high therapeutic dose may be reduced in a serial, step-wise fashion over a period of about 1 week to a level typically used in treating adrenal insufficiency in periods of crisis or stress, termed the stress physiologic dose.
Further tapering may then proceed during the next week, until a maintenance level is achieved at the presumed physiologic dose (basal levels that the body produces). Final tapering leading to discontinuation of therapy should then proceed slowly so that the duration of tapering approximates the total length of treatment, up to 6 to 9 months (Orth & Kovacs, 1998). Intercurrent febrile illness or surgical procedures demand a temporary increase in steroid dose for several days (Lamberts, Bruining, & de Jong, 1997).
Further tapering may then proceed during the next week, until a maintenance level is achieved at the presumed physiologic dose (basal levels that the body produces). Final tapering leading to discontinuation of therapy should then proceed slowly so that the duration of tapering approximates the total length of treatment, up to 6 to 9 months (Orth & Kovacs, 1998). Intercurrent febrile illness or surgical procedures demand a temporary increase in steroid dose for several days (Lamberts, Bruining, & de Jong, 1997).
Documentation of an intact HPA axis should be obtained before subjecting a patient to surgery who has a known history of prior high-dose, long-term glucocorticoid treatment. This may be done by documenting an 0800 plasma cortisol greater than 10 μg/dL or by performing low-dose cosyntropin challenge (see following section). If such documentation cannot be obtained in time, it is safest to treat with supplemental stress physiologic steroid coverage in the perioperative period for any patient within 1 year of withdrawal of steroid therapy (Oelkers, 1996). Figure 47-2 illustrates an algorithm for the tapering of long-term steroids.
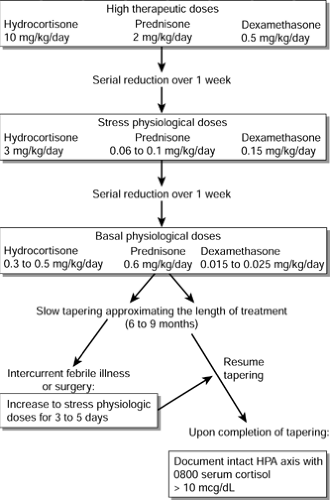 Figure 47-2 Tapering of long-term steroids. |
Although theoretically possible, adrenal insufficiency is uncommon in infants born to mothers treated with glucocorticoids during pregnancy. Only dexamethasone crosses the placenta to an appreciable degree.
Adrenal insufficiency is a common cause of mortality in the postoperative period for patients after pituitary surgery (Bates, Van’t Hoff, Jones, & Clayton, 1996). Patients who have been subjected to pituitary surgery should be covered with stress doses of glucocorticoids during and immediately after surgery. Several months after steroid tapering has been completed, such patients should be tested to determine whether the HPA axis is intact.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) should be considered in the differential diagnosis of adrenal insufficiency, especially
in the infant. Patients with CAH due to 21-hydroxylase deficiency (90%–95% of all CAH) cannot adequately synthesize cortisol. Inefficient cortisol synthesis causes the hypothalamus and pituitary to increase CRH and corticotropin (ACTH), respectively, and causes the adrenal glands to become hyperplastic. Instead of producing cortisol, the adrenals of CAH patients produce excess sex hormone precursors, which do not require 21-hydroxylation for their synthesis. Once secreted, these hormones are further metabolized to testosterone and dihydrotestosterone, active androgens, and to a lesser extent, estrogens, estrone, and estradiol. The net effect is prenatal virilization of the female fetus, producing a newborn girl with ambiguous genitalia. Males with CAH have no genital ambiguity. Rapid somatic growth with early epiphyseal fusion occurs in both sexes if the disease is not recognized and treated (Speiser & White, 1998).
in the infant. Patients with CAH due to 21-hydroxylase deficiency (90%–95% of all CAH) cannot adequately synthesize cortisol. Inefficient cortisol synthesis causes the hypothalamus and pituitary to increase CRH and corticotropin (ACTH), respectively, and causes the adrenal glands to become hyperplastic. Instead of producing cortisol, the adrenals of CAH patients produce excess sex hormone precursors, which do not require 21-hydroxylation for their synthesis. Once secreted, these hormones are further metabolized to testosterone and dihydrotestosterone, active androgens, and to a lesser extent, estrogens, estrone, and estradiol. The net effect is prenatal virilization of the female fetus, producing a newborn girl with ambiguous genitalia. Males with CAH have no genital ambiguity. Rapid somatic growth with early epiphyseal fusion occurs in both sexes if the disease is not recognized and treated (Speiser & White, 1998).
About 75% of patients also have insufficient aldosterone to maintain sodium balance and are termed salt wasters. These patients usually are diagnosed at 1 to 4 weeks of age with severe illness characterized by hyponatremia (serum sodium often < 120 mEq/L), hyperkalemia (serum potassium often > 7 mEq/L), acidosis, and hypovolemic shock. These adrenal crises may prove fatal if proper medical care is not delivered.
This problem is particularly critical in infant boys, who have no genital ambiguity to alert providers to the diagnosis of CAH prior to the onset of dehydration and shock.
Clinical clues to the diagnosis of salt-wasting CAH include lethargy, weak feeding pattern, vomiting, poor weight gain, pallor, or gray color with possible hyperpigmented scrotum and nipples.
Electrolytes should be measured in any infant with these features. The mortality rate for CAH remains high in such patients, suggested by the discrepancy in the number of male patients identified in case reports versus newborn screening. For this reason, many localities and several westernized foreign countries have included hormonal testing for CAH in the mandatory newborn disease screen (Pang & Shook, 1997).
Addison’s Disease
The most common cause of Addison’s disease is autoimmune (Laureti, Vecchi, Santeusanio, & Falorni, 1999). Type I polyglandular autoimmune syndrome, also termed Schmidt’s disease, has a mean age of presentation of 12 years. Hypoparathyroidism and adrenal insufficiency are the most frequent presenting signs, found in 79% and 72% of cases, respectively (Kong & Jeffcoate, 1994). Candidiasis, due to a defect in T cells’ handling of fungi, is the presenting sign of the disease in 60% of patients and is eventually detected in long-term follow-up of all patients. Antibodies can be detected to the adrenocortical steroidogenic enzymes, frequently to steroid 21-hydroxylase (Yu et al., 1999). Hypogonadism is seen more often in affected females than males. Other associated glandular defects include hypothyroidism and diabetes mellitus. Vitiligo is another cutaneous manifestation, while pernicious anemia and chronic active hepatitis are other systemic manifestations of this potentially fatal disease (Betterle, Greggio, & Volpato, 1998).
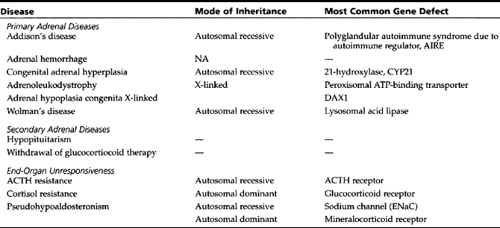
Tuberculosis and human immunodeficiency virus infection remain prominent causes of adrenocortical failure in children. Additional rare causes of adrenal insufficiency are listed in Table 47-2.
 |
Adrenal Hemorrhage
Unilateral adrenal hemorrhage is often asymptomatic; however, bilateral adrenal hemorrhage presents with shock due to subcapsular blood loss and adrenal insufficiency. Neonates may suffer adrenal hemorrhage after long labor and traumatic delivery with attendant hypoxemia. Acute and overwhelming infection due to meningococcemia (Waterhouse-Friderichsen syndrome), Haemophilus influenzae type B (no longer prevalent due to vaccination), group A β-hemolytic Streptococcus, or Pneumococcus may also be associated with bilateral adrenal hemorrhage and adrenal crisis. Prompt glucocorticoid administration can improve the outlook for these patients. Adrenal hemorrhage should be considered in blunt abdominal injury; in adrenal tumors, such as neuroblastoma and pheochromocytoma; and in patients with congenital or acquired bleeding diatheses or coagulation defects (Rao, 1995).
EPIDEMIOLOGY
The exact overall incidence of adrenal insufficiency in childhood is unknown because of its multifactorial etiology. Secondary adrenal insufficiency due to pituitary failure is probably the most common cause and is a frequent cause of morbidity and mortality in children who have undergone treatment with cranial radiation, surgery, or exogenous glucocorticoids for extended periods. No data are available on
the prevalence and incidence of pituitary-induced adrenal failure. The epidemiology of classic CAH due to 21-hydroxylase deficiency (21-OHD) is approximately 1 in 10,000 to 1 in 15,000 live births (Pang & Shook, 1997). Primary adrenal insufficiency, Addison’s disease, is relatively rare in the pediatric population. The prevalence in Great Britain is 110 per million population of all ages, with more than 90% of cases attributed to autoimmune destruction of the adrenal cortex. A similar prevalence was found in Umbria, Italy (Laureti et al., 1999). The calculated incidence is approximately 5 to 6 per million population per year (Kong & Jeffcoate, 1994).
the prevalence and incidence of pituitary-induced adrenal failure. The epidemiology of classic CAH due to 21-hydroxylase deficiency (21-OHD) is approximately 1 in 10,000 to 1 in 15,000 live births (Pang & Shook, 1997). Primary adrenal insufficiency, Addison’s disease, is relatively rare in the pediatric population. The prevalence in Great Britain is 110 per million population of all ages, with more than 90% of cases attributed to autoimmune destruction of the adrenal cortex. A similar prevalence was found in Umbria, Italy (Laureti et al., 1999). The calculated incidence is approximately 5 to 6 per million population per year (Kong & Jeffcoate, 1994).
HISTORY AND PHYSICAL EXAMINATION
Clinical clues to the diagnosis of adrenal insufficiency are extremely nonspecific. Infants may be jittery or have convulsions due to hypoglycemia. Chronic adrenal insufficiency will result in failure to thrive. Older children may exhibit fatigue, malaise, muscle aches, headache, abdominal pain, nausea, vomiting, salt craving, and weight loss with glucocorticoid or mineralocorticoid deficiencies.
Physical examination can provide important clues that direct the clinician to appropriate laboratory diagnosis. In suspected adrenal insufficiency, as in the evaluation of any chronic childhood illness, careful review of linear growth and weight gain is critical to understanding the nature and chronicity of the problem.
• Clinical Pearls:
Orthostatic hypotension and pulse changes are sensitive bedside indications of mineralocorticoid deficiency or volume depletion.
In untreated primary adrenal insufficiency, physical examination often shows hyperpigmentation, especially of non–sun-exposed areas, such as palms, soles, and oral mucosa.
This is a direct result of cosecretion of melanocyte-stimulating hormone with pro-opiomelanocortin, the precursor of ACTH. In the case of adrenal hemorrhage or tumor, a flank mass may be palpated on careful abdominal examination. Patients with global adrenocortical insufficiency chronically lacking adrenal androgens will often have sparse pubic and axillary hair. On the other hand, CAH patients produce excess androgens.
• Clinical Pearls:
Females with CAH will show genital ambiguity as neonates, and girls with CAH not detected in infancy often have an enlarged clitoris without true genital ambiguity.
Males with CAH are much more difficult to identify clinically. They may show an enlarged penis and pubic hair and a hyperpigmented scrotum with small testes.
DIAGNOSTIC CRITERIA AND STUDIES
Relevant basic laboratory studies to confirm adrenal insufficiency include serum and urine electrolytes, glucose, early morning cortisol, ACTH, and plasma renin activity. One expects to find low serum (and high urine) sodium, high serum (and low urine) potassium with metabolic acidosis. There is an inappropriately low cortisol for the degree of ACTH elevation and high plasma renin activity. One should not attempt to restrict sodium intake in a patient with adrenal insufficiency, except under direct medical supervision, because this can provoke adrenal crisis. If central adrenal insufficiency is suspected, a low-dose cosyntropin stimulation test (1 μg intravenous [IV] bolus of ACTH 1-24) is the best way to assess the intactness of the HPA axis. Cortisol should rise after 30 to 60 minutes to a level greater than 15 to 20 μg/dL (Oelkers, 1996).
• Clinical Pearl
If CAH is suspected, the primary care provider should at least obtain a rapid measurement of basal serum 17-hydroxyprogesterone by radioimmunoassay.
Classic, severely affected CAH patients usually have markedly elevated random basal serum 17-hydroxyprogesterones
in the range of greater than 10,000 to 20,000 ng/dL (New et al., 1983). Newborn screening programs are in effect in several states and rely primarily on filter paper determinations of blood 17-hydroxyprogesterone. The cost, sensitivity, specificity, and predictive values of these screening tests vary depending on the assay, the number of tests, and the established cut-off levels. Premature infants tend to have higher levels of 17-OHP than term infants and generate many false-positive results. Suggested weight-adjusted cut-offs range from 165 ng/mL in blood collected on filter paper for infants under 1300 g to 40 ng/mL for infants over 2200 g in Wisconsin (Allen et al., 1997); in Texas, cut-offs of 40 and 65 ng/mL are used for infants greater or less than 2500 g, respectively (Therrell et al., 1998). Because some radioimmunoassays cross-react with other steroids, positive screens should be confirmed. A more specific diagnostic test involving a complete profile of adrenocortical hormones before and 1 hour after high-dose cosyntropin stimulation (250 μg IV bolus) is obtained to distinguish CAH due to 21-hydroxylase deficiency from other forms of adrenal disease.
in the range of greater than 10,000 to 20,000 ng/dL (New et al., 1983). Newborn screening programs are in effect in several states and rely primarily on filter paper determinations of blood 17-hydroxyprogesterone. The cost, sensitivity, specificity, and predictive values of these screening tests vary depending on the assay, the number of tests, and the established cut-off levels. Premature infants tend to have higher levels of 17-OHP than term infants and generate many false-positive results. Suggested weight-adjusted cut-offs range from 165 ng/mL in blood collected on filter paper for infants under 1300 g to 40 ng/mL for infants over 2200 g in Wisconsin (Allen et al., 1997); in Texas, cut-offs of 40 and 65 ng/mL are used for infants greater or less than 2500 g, respectively (Therrell et al., 1998). Because some radioimmunoassays cross-react with other steroids, positive screens should be confirmed. A more specific diagnostic test involving a complete profile of adrenocortical hormones before and 1 hour after high-dose cosyntropin stimulation (250 μg IV bolus) is obtained to distinguish CAH due to 21-hydroxylase deficiency from other forms of adrenal disease.
Radiographic imaging can be a helpful adjunct to biochemical testing in some types of adrenal insufficiency. Ultrasound is used in neonates with suspected adrenal hemorrhage; adrenal enlargement and inferior renal displacement may be detected. If done at 5-day intervals, ultrasound imaging may document changes in the lesion from solid to cystic to final calcification about a month later. Computed tomographic (CT) scan or magnetic resonance imaging is most sensitive for imaging adrenal pathology in older patients (Brant, 1999).
MANAGEMENT
Prompt recognition of adrenal insufficiency is the key to a good outcome. Timely initiation of glucocorticoid replacement therapy is the most important factor.
• Clinical Pearl
When acute adrenal failure is recognized, an initial bolus of parenteral hydrocortisone (25 mg for infants, 50 mg for children, or 100 mg for adolescents and adults) of hydrocortisone sodium succinate (Solu-Cortef) either IV or intramuscular [IM]) can be life-saving.
Hydrocortisone has mineralocorticoid properties, and thus additional mineralocorticoid treatment is not immediately necessary. Drugs such as prednisone and dexamethasone do not possess significant mineralocorticoid activity and are not first-choice drugs for acute treatment (see Table 47-1). Because all patients in adrenal crisis and many with chronic adrenal insufficiency are dehydrated, isotonic fluid administration, in the form of a 20 mL/kg bolus of IV normal saline or lactated Ringer’s solution, is also required. Subsequent fluid replacement should be tailored to the individual patient’s needs, based on blood pressure, heart rate, mental status, and capillary refill.
Continued steroid treatment should be ordered for the acutely ill patient as IV hydrocortisone every 6 hours, at 3 mg/kg per day for the first 24 hours, representing a stress physiologic dose. Subsequently, the dose can be tapered until a reasonable maintenance dose is achieved. In children, the preferred maintenance cortisol replacement is hydrocortisone (which is the same as cortisol) in two or three divided doses totaling 0.3 mg/kg per day (or about 10 mg/m2 per day), the basal physiologic dose. CAH patients often require higher doses of glucocorticoids, not to exceed 0.6 mg/kg per day (or about 20 mg/m2 per day). A minimum dose of 6 mg/d of hydrocortisone (oral hydrocortisone cypionate, Cortef, suspension 10 mg/5 mL) in three divided doses is usually administered to infants with CAH.
The short half-life of hydrocortisone minimizes growth suppression and other adverse side effects of longer acting, more potent glucocorticoids, such as prednisone and dexamethasone. However, a single daily dose of a short-acting glucocorticoid is insufficient as replacement therapy and therefore a longer-acting steroid may be used in patients who cannot comply with a drug regimen requiring frequent dosing.
Efficacy of Treatment
Treatment efficacy for Addison’s disease cannot be assessed by measuring plasma cortisol, because only exogenous hydrocortisone would be measured. Plasma ACTH is also not a reliable measure of treatment efficacy, because this rather labile hormone has circadian variability. Rather, the patient’s sense of well-being is most important. Symptoms such as chronic headache, dizziness, and fatigue would suggest insufficient treatment.
Treatment efficacy for CAH (ie, suppression of adrenal sex hormones) is monitored by 17-OHP and androstenedione levels. Testosterone can also be a useful parameter in females and prepubertal males. The target 17-OHP range is usually 100 to 1000 ng/dL, depending on age and sex. Hormones should be measured consistently at the same time in relation to medication dosing. Plasma renin activity, blood pressure, and pulse are useful measures of sodium and fluid repletion.
Side Effects
Overtreatment with glucocorticoids reduces linear growth and promotes centripetal obesity, hyperglycemia, hypertension, and bone mineral loss, as well as psychologic sequelae ranging from depression to psychosis. It is therefore advisable to evaluate steroid-treated children three to four times annually to assess linear growth, weight gain, and blood pressure.
Adults should be monitored at least yearly. As noninvasive techniques for measuring bone density become more widely available, these tests may eventually become standard for such patients.
What to Tell Parents
There are both transient and permanent forms of adrenal insufficiency. In either case, it is important to stress that steroid treatment and hydration are vital to support the patient at risk for adrenal crisis during surgery or other potentially life-threatening periods. Individuals diagnosed with permanent forms of adrenal insufficiency (Addison’s disease and CAH) should wear a medallion indicating their diagnosis and medications. Primary care practitioners and school health personnel should be informed of necessary measures in case of shock. A home kit with injectable hydrocortisone sodium succinate should be supplied, and parents or caregivers instructed in its proper use. A person with adrenal insufficiency who is either lethargic or unresponsive outside the hospital setting should be given an IM injection of hydrocortisone sodium succinate (25 mg infant, 50 mg child, 100 mg adult). An IV line with isotonic fluids should be established and the patient brought promptly to an acute care facility. If the individual is conscious, clear electrolyte
fluids should be provided (sports drinks, such as Gatorade or Powerade, are ideal).
fluids should be provided (sports drinks, such as Gatorade or Powerade, are ideal).
Addison’s disease and CAH are unremitting conditions requiring lifelong treatment. Nevertheless, most patients lead unrestricted and productive lives. Patients receiving low-dose maintenance glucocorticoid therapy are not at risk for immunosuppression. There are no significant interactions between these medications and other commonly used pediatric drugs.
Referral
Most primary care providers will request consultation from an endocrinologist to help care for patients with known or suspected adrenal insufficiency. The clinician should be familiar with signs and symptoms of these disorders, and should perhaps discuss basic laboratory evaluation before referring the patient to the specialist. It is particularly important to recognize signs of systemic progression of polyglandular endocrinopathy, and such patients should undergo annual screening for other glandular malfunctions specified above.
COMMUNITY RESOURCES
The following organizations provide information and support to patients and families with various adrenal disorders.
Magic Foundation
1327 N. Harlem Avenue
Oak Park, IL 60302
Telephone: (708) 383-0808
Fax: (708) 383-0899
Internet address: http://www.magicfoundation.org
This foundation for children and adults with growth disorders offers educational brochures, a networking system for parents, newsletters for adults and children, and divisions on specific disorders.?
National Adrenal Diseases Foundation
505 Northern Boulevard
Great Neck, NY 11021
Telephone: (516) 487-4992
Internet address: http://www.medhelp.org/nadf
National Institutes of Health
Pituitary Tumor Network Association
Telephone: (805) 499-2262,
Internet address: http://www.pituitary.com
REFERENCES
Allen, D. B., Hoffmann, G. L., Fitzpatrick, P., Laessig, R., Maby, S., & Slyper, A. (1997). Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. Journal of Pediatrics, 130, 128–133.
Bates, A. S., Van’t Hoff, W., Jones, P. J., & Clayton, R. N. (1996). The effect of hypopituitarism on life expectancy. Journal of Clinical Endocrinology and Metabolism, 81, 1169–1172.
Betterle, C., Greggio, N. A., & Volpato, M. (1998). Clinical review 93: Autoimmune polyglandular syndrome type 1. Journal of Clinical Endocrinology and Metabolism, 83, 1049–1055.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


