INTRODUCTION
Controversy has existed concerning acid-base physiology and the ideal method to assess acid-base disorders for 130 years.1 The two most common methods advocated to analyze acid-base disorders are the traditional bicarbonate-centered method2,3 and the Stewart, or strong ion, method.4,5 The traditional approach teaches that acid-base homeostasis is maintained by respiratory control of the partial pressure of carbon dioxide (Pco2) through changes in alveolar ventilation and control of HCO3– reabsorption and H+ excretion by the kidneys. Peter Stewart proposed that acid-base physiology involves the dynamic interaction of body fluids and multiple chemical species including strong ions (primarily Na+, K+, Ca2+, Mg2+, and Cl–) and weak acids, as well as Pco2 control by the lungs.
Each of these methods has limitations. The traditional bicarbonate-centered model continues to be the most commonly used at the bedside3 but is criticized for failing to identify acid-base abnormalities that are due to alterations in plasma free water or in complex cases of mixed acid-base disorders.6,7 The Stewart method is praised for its accuracy in identifying acid-base disorders but is criticized for the difficulty of application at the bedside.7,8,9,10,11 This chapter does not detail the Stewart method, but we acknowledge its importance and its contribution to underscoring the limitations of the traditional method, which has led to modifications that improve the performance of the traditional method at the bedside.8 For example, using a correction factor for the albumin level (detailed in this chapter), the traditional method performs as well as the Stewart method for identifying complex acid-base abnormalities in critically ill patients.8,9,10,11,12
Many diseases, including those that present an imminent threat to life, produce acid and base (acid-base) disturbances that provide important clues concerning the nature of the underlying illness and suggest immediate therapeutic interventions. Further, ED treatments such as rapid resuscitation of critically ill patients may create unintended acid-base disorders. This chapter describes a practical approach to the clinical evaluation and treatment of acid-base disorders.
PATHOPHYSIOLOGY
Plasma hydrogen ion concentration ([H+])* is normally 40 nmol/L, corresponding to a pH of 7.4. Because pH is a logarithmic transformation of [H+], the relation of [H+] to pH is not linear for all pH values (Table 15-1). However, for pH values from 7.20 to 7.50, the relation between [H+] and pH is nearly linear; pH changes of 0.01 correspond to approximately 1 nmol/L change in [H+]. This linear relation allows for rapid bedside interpretation of blood gas and electrolyte results.
*Standard nomenclature is used in this chapter. The presence of brackets, [ ], surrounding an element or molecule implies the term concentration. Without the brackets, the chemical expressions simply refer to the element or molecule.
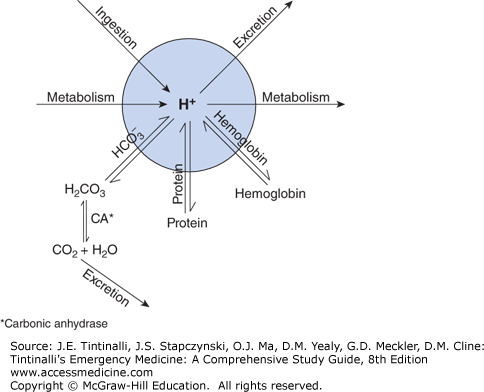
Plasma [H+] is influenced by the rate of endogenous production, the rate of excretion, exogenous addition (e.g., acetylsalicylic acid ingestion), and the buffering capacity of the body. Buffers mitigate the impact of large changes in available hydrogen ion on plasma pH.
Buffer systems that are effective at physiologic pH include hemoglobin, phosphate, proteins, and bicarbonate (Figure 15-1). One can consider the [H+] to be the result of all physiologic buffers acting on the common pool of hydrogen ions.
The familiar Henderson-Hasselbalch equation, shown in Eq. (1),
specifies the relationship between carbonic acid, bicarbonate, and pH; any two of these determine the value of the third. The clinical use of the Henderson-Hasselbalch equation is limited. However, if all constants are inserted into the Henderson-Hasselbalch equation and the anti-logarithm of all its terms is taken, the result is the Kassirer-Bleich equation [Eq. (2)], which is of great clinical utility.
The Kassirer-Bleich equation may be used to estimate the concentration of any component of the bicarbonate buffer system provided the concentrations of the other two components are known. Therefore, it allows clinicians to determine, for example, what the pH must be when the Pco2 and [HCO3–] are known.† Note that when [HCO3–] is normal, the Kassirer-Bleich equation demonstrates that [H+] = Pco2, using their respective units of measure.
The quantity of [HCO3–] in relation to carbonic acid buffer in the system is not fixed, but varies according to physiologic need. This flexibility is largely provided by pulmonary exhalation of carbon dioxide (CO2), which can vary significantly and change rapidly as required by alterations in the underlying acid-base status.
The kidney regulates HCO3– excretion and the formation of new HCO3–. The rate of these processes is dependent on the underlying acid-base status. The renal response to pulmonary acid-base disturbances begins within 30 minutes of onset, but requires hours to days to achieve equilibrium.13 Bicarbonate is filtered into the urine and must be reclaimed to maintain homeostasis. Eighty-five percent of bicarbonate that is filtered is reclaimed by the proximal convoluted tubule in a sodium-dependent process. H+ is secreted into the tubular lumen, and Na+ is absorbed from the tubular lumen into the cell. Sodium is then extruded from the tubular cell into the plasma in exchange for K+ via the Na+/K+-ATPase pump. Thus, the H+ secreted into the tubule combines with the filtered HCO3– in the lumen to produce carbonic acid (H2CO3), which in turn is converted to CO2 and H2O by carbonic anhydrase. The CO2 diffuses down its concentration gradient into the tubule cell, where cytoplasmic carbonic anhydrase regenerates H2CO3, which then dissociates into HCO3– and H+ (creating a supply of H+ for extrusion). If tubular disease inhibits H+ extrusion, a proximal renal tubular acidosis results, in which serum [HCO3–] decreases to a steady-state level, i.e., reclaimed HCO3– effectively equals H+ extrusion.
The balance (15%) of HCO3– reclamation occurs in the distal tubule via a sodium-independent process. Cytoplasmic carbonic anhydrase generates H2CO3–, which dissociates. HCO3– diffuses into plasma, and H+ is secreted into the lumen by an H+-ATPase pump, thereby maintaining cellular electrical neutrality. H+ is trapped in the lumen by inorganic phosphate or ammonia (NH4+). Failure of H+ secretion is the underlying mechanism of distal renal tubular acidosis.
New HCO3– can also be created by the kidney. A sodium-dependent process allows synthesis of HCO3– in the distal tubule. Intracellular glutamine generates HCO3– and ammonia NH4+. The Na+/K+ pump moves Na+ into the lumen, but Na+ diffuses back across the cell membrane, and NH4+ is secreted into the lumen in exchange. The generated HCO3– remains in the cell. Formation of HCO3– by this process increases during acidosis, but may require 4 to 5 days to reach equilibrium. Drugs that alter uptake or delivery of Na+ to the distal tubule can significantly alter HCO3– synthesis. The process of acid secretion allows the regeneration of HCO3– in proportion to the daily production of acid. Urine, especially under conditions of acidosis, can be made almost entirely without HCO3–.
†The “bicarbonate concentration” measured by the clinical laboratory is actually the total CO2, which is the sum of bicarbonate, dissolved CO2, and H2CO3. H2CO3 is the Pco2 multiplied by the solubility coefficient of CO2 in blood (α), 0.03. Thus, total CO2 = [HCO3–] + (0.03) (Pco2). Most clinicians simply neglect the second term when the Pco2 is normal; when hypercapnia is present, however, the second term measurably contributes to total CO2.
FUNDAMENTAL ACID-BASE DISORDERS
Any condition that acts to increase [H+]—whether through endogenous production, decreased buffering capacity, decreased excretion, or exogenous addition—is known as acidosis. Similarly, any condition that acts to decrease [H+] is termed alkalosis. The terms acidemia and alkalemia refer to the net imbalance of [H+] in the blood. The difference between acidosis and acidemia is not merely semantic, but of great clinical importance. For example, a patient with acidosis and alkalosis of equal magnitude will have a normal pH. A patient with these disturbances thus has neither acidemia nor alkalemia (resulting in the normal pH), but nevertheless has both acidosis and alkalosis. It is important to appreciate that, although acidemia is diagnostic of acidosis and alkalemia of alkalosis, a normal or high pH does not exclude acidosis and a normal or low pH does not exclude alkalosis.
Acid-base disturbances are further classified as respiratory or metabolic. Respiratory acid-base disorders are due to primary changes in Pco2, and metabolic acid-base disorders reflect primary changes in [HCO3–]. Compensatory mechanisms are, by definition, not “disorders,” but rather normal physiologic responses to acid-base derangements. Terms such as compensatory respiratory alkalosis are therefore misleading. The clinician is nonetheless concerned with the adequacy of compensation, because failure of appropriate compensatory response implies the presence of another primary acid-base disturbance.
It is important to note that compensatory mechanisms return the pH toward normal but do not reach baseline.‡ The fact that compensatory mechanisms cannot reach completion is evident when one considers that complete compensation would necessarily remove the physiologic stimulus driving the compensation.2
The “normal” values of pH, Pco2, and [HCO3–] for given laboratory ranges are intended to include 95% of patients without an acid-base disorder. The normal pH range is 7.35 to 7.45, the normal Pco2 range is 35 to 45 mm Hg, and the normal [HCO3–] is usually 21 to 28 mEq/L. However, a patient’s values may all fall within the “normal range” and still have significant acid-base disturbances. As detailed further below, a patient with metabolic acidosis and a concomitant metabolic alkalosis of nearly approximate magnitude will have a “normal” pH, Pco2, and [HCO3–]. In contrast, abnormal values may be appropriate for a given simple acid-base disturbance. For example, in the presence of a metabolic acidosis where [HCO3–] = 15 and pH = 7.3, an appropriate respiratory compensation should result in a Pco2 of about 30 mm Hg. This Pco2 value is below the “normal” range, but at the expected level of physiologic respiratory compensation for the degree of metabolic acidosis. In this example, the finding of Pco2 in the normal (35 to 45 mm Hg) range actually implies the presence of a respiratory acidosis, because the expected physiologic respiratory response is inadequate.
The principle of electrical neutrality requires that plasma have no net charge. The charge of the predominant plasma cation, Na+, must therefore be “balanced” by the charge of plasma anions. Although HCO3– and Cl– constitute a significant fraction of plasma anions, the sum of their concentrations does not equal that of sodium. Therefore, there must be other anions present in the serum to maintain electrical neutrality. These anions are primarily serum proteins (albumin, phosphate, sulfate, and organic anions, such as lactate) and the conjugate bases of ketoacids. Because these substances are not commonly measured, they are termed unmeasured anions. Unmeasured cations also exist, largely in the form of Ca2+ and Mg2+. Because all cations (measured cations [MC] and unmeasured cations [UC]) must equal all anions (measured anions [MA] and unmeasured anions [UA]):
it follows that:
Thus, substituting measured ions produces:
The unmeasured anion concentration is commonly called the anion gap (AG), i.e., the difference between the serum [Na+] and the sum of serum [Cl–] and [HCO3–] equals the concentration of the unmeasured anions. The contribution of [K+], largely an intracellular ion, is usually neglected (although some hospital laboratories still include the [K+] as part of the reported AG value). For the purposes of AG determination, correction of serum [Na+] in the face of hyperglycemia is unnecessary, because this condition similarly dilutes [Cl–]. The normal value of the AG is generally considered to be 12 ± 4 mEq/L, assuming no major deviations in expected concentration of unmeasured anions or cations. Reports have suggested that a normal AG value of 7 ± 4 mEq/L may be more appropriate to electrolyte measurements made with ion-specific electrodes.14 However, normal range values used by the clinician should reflect institutional practice. As with other acid-base concepts, the accepted “normal” range for the AG is less important than whether it has changed in relation to the patient’s steady-state baseline value. Thus, a relative change in the AG, referred to as the delta gap, may be more important than the actual AG value. Virtually all AG values above 15 mEq/L can be considered abnormal, even when there are no previous comparison values available.
The AG may change even in the absence of acid-base disturbances. It may rise when (unmeasured) cations decrease, as in severe states of hypomagnesemia, hypokalemia, and hypocalcemia. A reduced, narrow, or even negative AG may result from an increase in the concentration of unmeasured cations, such as lithium; unmeasured positively charged proteins resulting from myeloma and polyclonal gammopathies; or a significant decrease in unmeasured anions, such as albumin and γ-globulin. Albumin is a major component of the AG. Critically ill patients are frequently hypoalbuminemic, which may decrease the AG into the normal range, effectively masking the presence of a wide AG acidosis.15 The AG should be corrected [AG-Corr(albumin)]6,8 for an abnormal albumin level to improve the sensitivity of using the AG to identify a metabolic acidosis:#
A factitiously narrow or even negative AG may result from a number of conditions including measurement artifact. Bromide toxicity yields false elevations of chloride, unless Cl– specific electrodes are used for detection;** triglyceride levels greater than 600 mg/dL falsely elevate chloride levels and lower sodium levels, resulting in an apparently narrow or even negative AG.16 If these can be excluded, a narrow AG may imply an excess of unmeasured cations such as may occur with hypergammaglobulinemias and myeloma proteins.
Although increases in the AG are traditionally considered in the context of metabolic acidosis, elevation of the AG may be seen with other acid-base disturbances. Metabolic and respiratory alkalosis, for example, may elevate the AG by 2 to 3 mEq/L, due to elevations in lactate (an unmeasured anion) produced by enhancement of glycolysis.†† Penicillin and carbenicillin, as unmeasured anions, produce elevations in the AG and may be accompanied by a hypokalemic alkalosis.
Elevations of the AG are usually clinically importance in the emergency setting and most commonly associated with metabolic acidosis (Table 15-2). Traditional mnemonics for the differential diagnosis of an elevated AG acidosis (MUDPILES, CAT MUDPILES, GOLDMARK, KARMEL, KUPIN, ACE GIFTs) can help the clinician recall elements of the differential diagnosis for this condition. However, these mnemonics leave the dangerous impression that lactic acidosis is a diagnostic endpoint for elevated AG acidosis. It is not. For example, propylene glycol, iron, seizures from isoniazid, carbon monoxide poisoning, and aspirin ingestions, as well as alcoholic ketoacidosis, may produce significant lactic acidosis. We suggest that the differential diagnosis of metabolic acidosis with an elevated AG should emphasize distinctions between endogenous and exogenous unmeasured anion sources and avoid mixing the etiology of lactic acidosis with that of other increased unmeasured anions. Some authors suggest correcting for lactate in patients known to have lactate elevations at baseline;17,18 if lactate correction is calculated, the delta lactate should be used.
| Diagnostic Category | Anion Species | Origin | Diagnostic Adjuncts |
|---|---|---|---|
| Renal failure (uremia) | [PO42–], [SO42–] | Protein metabolism | BUN/creatinine |
| Ketoacidosis | Ketoacids, lactate | Fatty acid metabolism | Serum/urine ketones |
| Diabetic | β-Hydroxybutyrate, lactate | Fatty acid metabolism | Specific test now available (older nitroprusside test yields false-negative result for β-hydroxybutyrate) |
| Alcoholic | Acetoacetate, lactate | Fatty acid metabolism | |
| Starvation | Consider coexistent dehydration | ||
| Lactic acidosis* | Lactate | Metabolism | Lactate level for subtypes |
| Sepsis | Lactate | Hypoperfusion, anaerobic metabolism | Culture/organism-specific tests |
| Cardiac arrest | Lactate | Hypoperfusion/reperfusion injury | Consider other acidosis |
| Liver failure | Lactate | Decreased lactate clearance | Liver function tests |
| Iron | Lactate | Disruption of cellular metabolism, | Serum iron level |
| Metformin | Lactate | Inhibition of gluconeogenesis | |
| Cyanide | Lactate | Mitochondrial dysfunction, histotoxic hypoxia | |
| Carbon monoxide | Lactate | Hypoxia, anaerobic metabolism | Carbon monoxide level |
| Thiamine deficiency | Lactate | Aerobic metabolism interrupted, lactate accumulates | Assess peripheral sensory and motor function for neuropathy |
| Exogenous poisoning* | |||
| Methanol | Formate | Methanol metabolism | Osmolal gap |
| Ethylene glycol (EG) | Oxalate and organic anions | EG metabolism favors pyruvate conversion to lactate | Osmolal gap Oxalate crystals (urine) |
| Salicylate | Salicylate | Salicylate, lactate, ketoacids | Concomitant respiratory alkalosis and metabolic acidosis |
| Isoniazid | Lactate | Anaerobic metabolism, lactate accumulation |
Clinical use of AG values requires an appreciation of their limitations. Although an AG greater than 30 mEq/L is usually caused by lactic acidosis or diabetic ketoacidosis, these conditions may exist even when the AG is normal.19 Thus, a “normal” AG does not exclude the possibility of the presence of increased concentrations of unmeasured cations. An AG increased from baseline but still within the “normal” range may be a clue (delta gap). Direct measurements of lactate, formate (parent of formic acid), ketoacids, methanol, ethylene glycol (parent of oxalic acid and numerous other organic acids), and salicylate should be ordered when the presence of any of these substances is suspected, but the AG is “normal,” (when a delta gap exists, or the AG is in the upper range of normal). Measurement of serum osmolarity and subsequent comparison to calculated serum osmolarity are necessary to detect small unmeasured molecules (such as toxic alcohols).20 Finally, it is important to recognize that several concomitant causes of wide AG–type metabolic acidosis may be present.
A common clinical problem is the diagnosis of mixed acid-base disturbances in the presence of an elevated AG. Simple acid-base disturbances that produce elevated AGs are referred to as wide AG metabolic acidoses. If a wide AG metabolic acidosis is the only disturbance, then the change (elevation from baseline) in value of the AG (the delta gap) should exactly equal the net decrease in the [HCO3–]. This is a one-to-one relationship. This concept is represented mathematically in Eq. (7).
If the [HCO3–] is lower than the value predicted by the delta AG, then there must be a concomitant hyperchloremic (i.e., normal AG type) metabolic acidosis (Figure 15-2A). Similarly, if the [HCO3–] is higher than expected based on the delta AG, there must be a concomitant metabolic alkalosis present. Note that acute respiratory conditions (respiratory acidosis or alkalosis) do not affect these determinations, although chronic respiratory conditions may have substantial metabolic compensatory effects. Potential acid-base disturbances related to respiratory status must be further determined, as discussed below (Figure 15-2A–C).
FIGURE 15-2.







