Decreased production/hypoproliferation
 Ineffective erythropoiesis
Ineffective erythropoiesis
 Decreased red cell survival
Decreased red cell survival
Common Causes to Remember
Epidemiology
 RBC lifespan ~120 days
RBC lifespan ~120 days
 Etiology of anemia in intensive care unit (ICU) patients is often multifactorial
Etiology of anemia in intensive care unit (ICU) patients is often multifactorial
 Incidence in ICU patients between 29% and 37%
Incidence in ICU patients between 29% and 37%
 44% of ICU patients in the United States will receive one blood transfusion during their ICU stay
44% of ICU patients in the United States will receive one blood transfusion during their ICU stay
Key Pathophysiology
 Hemodilution
Hemodilution
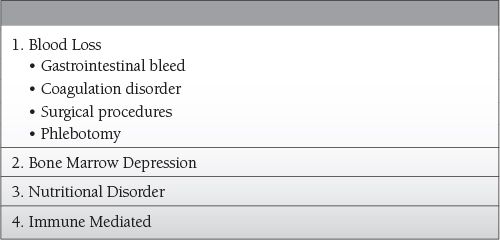
 Blood Loss
Blood Loss
 Phlebotomy
Phlebotomy
 Hemorrhage
Hemorrhage
 Reduced RBC survival and production
Reduced RBC survival and production
 Iron Metabolism
Iron Metabolism
 B12/Folate Metabolism
B12/Folate Metabolism
 Decreased Erythropoietin Concentration
Decreased Erythropoietin Concentration
 Abnormal Red Cell Maturation
Abnormal Red Cell Maturation
Anemia in the Critically Ill:
Critical Illness → Blood Loss and/or decreased production → Immune activation → Decreased Iron → Decreased erythropoiesis
Differential Diagnosis
 Iron Deficiency Anemia
Iron Deficiency Anemia
 Most common cause of anemia
Most common cause of anemia
 Patients with iron deficiency anemia have longer ICU stays
Patients with iron deficiency anemia have longer ICU stays
 Hemolytic Anemia (RBC destruction)
Hemolytic Anemia (RBC destruction)
 Intrinsic defect of the RBC; usually inherited (sickle cell anemia, thalassemias)
Intrinsic defect of the RBC; usually inherited (sickle cell anemia, thalassemias)
 Extrinsic (aka autoimmune hemolytic anemia)
Extrinsic (aka autoimmune hemolytic anemia)
 Infectious—hepatitis, CMV, EBV, Escherichia coli
Infectious—hepatitis, CMV, EBV, Escherichia coli
 Medications—PCN, sulfa, anti-malarials
Medications—PCN, sulfa, anti-malarials
 Diseases—SLE, rheumatoid arthritis, ulcerative colitis
Diseases—SLE, rheumatoid arthritis, ulcerative colitis
 Blood Loss
Blood Loss
Management and Treatment
(see Transfusion Therapy, Section 18)
 Blood Transfusions
Blood Transfusions
 Erythropoietin Therapy
Erythropoietin Therapy
SUGGESTED READINGS
Henry ML, Garner WL, Fabri PJ. Iatrogenic anemia. Am J Surg. 1986;151:362.
Shander A. Anemia in the critically ill. Crit Care Clin. 2004;159-170.
Vincent JL, Baron JF, Reinhart K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288:1499-1507.
VonAhsen N, Muller C, Serke S, Frei U, Eckardt KU. Important role of nondiagnostic blood loss and blunted erythropoietic response in the anemia of medical intensive care patients. Crit Care Med. 1999;27: 2630-2639.
Walsh TS, Saleh E. Anemia during critical illness. BJA. 2006;97:278-291.
7.2
Sheri M. Berg
Disease of decreased number of platelets characterized by
 Platelet count < 150,000
Platelet count < 150,000
 Petechiae
Petechiae
 Purpura
Purpura
 Bruising
Bruising
 Frank bleeding
Frank bleeding
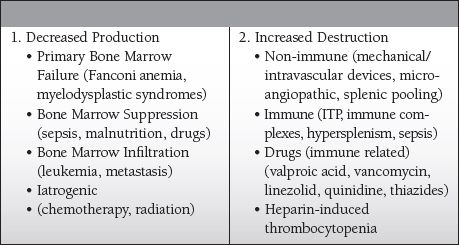
Common Causes to Remember
Epidemiology
 Platelet lifespan ~10 days
Platelet lifespan ~10 days
 Etiology of thrombocytopenia in intensive care unit (ICU) patients can be multifactorial
Etiology of thrombocytopenia in intensive care unit (ICU) patients can be multifactorial
 Most common causes in the ICU are drug exposure and sepsis
Most common causes in the ICU are drug exposure and sepsis
 Incidence in ICU patients between 13% and 60%
Incidence in ICU patients between 13% and 60%
 Associated with increased length of stay, morbidity, and mortality
Associated with increased length of stay, morbidity, and mortality
Key Pathophysiology
 Dilutional—transfusion of crystalloid, packed cells, and/or FFP without replacing platelets
Dilutional—transfusion of crystalloid, packed cells, and/or FFP without replacing platelets
 Distributional—splenic sequestration in those with splenomegaly
Distributional—splenic sequestration in those with splenomegaly
 Destruction—(+ schistocytes on blood smear, as there is usually associated hemolysis)
Destruction—(+ schistocytes on blood smear, as there is usually associated hemolysis)
 Nonimmune
Nonimmune
 Mechanical devices that cause shearing of cells, such as an IABP or LVAD
Mechanical devices that cause shearing of cells, such as an IABP or LVAD
 Thrombotic thrombocytopenic purpura (TTP)
Thrombotic thrombocytopenic purpura (TTP)
 Hemolytic uremic syndrome (HUS)
Hemolytic uremic syndrome (HUS)
 Immune
Immune
 Idiopathic thrombocytopenic purpura (ITP)
Idiopathic thrombocytopenic purpura (ITP)
 Heparin-induced thrombocytopenia (HIT)
Heparin-induced thrombocytopenia (HIT)
 Drug induced (can be immune mediated or nonimmune)
Drug induced (can be immune mediated or nonimmune)
Differential Diagnosis
 TTP
TTP
 Microangiopathic process with thrombocytopenia and microvascular thrombosis being the most prominent features
Microangiopathic process with thrombocytopenia and microvascular thrombosis being the most prominent features
 Platelet aggregates contain vonWillebrand factor antigen
Platelet aggregates contain vonWillebrand factor antigen
 Classic “pentad”: thrombocytopenia, microangiopathic hemolytic anemia, fever, mental status changes, and renal insufficiency
Classic “pentad”: thrombocytopenia, microangiopathic hemolytic anemia, fever, mental status changes, and renal insufficiency
 “Triad” of thrombocytopenia, schistocytosis, and elevated lactate dehydrogenase levels suggests TTP
“Triad” of thrombocytopenia, schistocytosis, and elevated lactate dehydrogenase levels suggests TTP
 HUS
HUS
 Hallmark = renal failure
Hallmark = renal failure
 Predominantly renal platelet-fibrin thrombi
Predominantly renal platelet-fibrin thrombi
 Often preceded by a gastrointestinal illness with a gram-negative cytotoxin-producing bacteria (Escherichia coli, Shigella)
Often preceded by a gastrointestinal illness with a gram-negative cytotoxin-producing bacteria (Escherichia coli, Shigella)
 ITP
ITP
 Autoimmune (platelets become coated with IgG antibodies)
Autoimmune (platelets become coated with IgG antibodies)
 Hallmark = low platelets and mucocutaneous bleeding
Hallmark = low platelets and mucocutaneous bleeding
 Often a diagnosis of exclusion
Often a diagnosis of exclusion
 HIT
HIT
 Autoimmune (IgG antibodies to the heparin–platelet factor IV complex)
Autoimmune (IgG antibodies to the heparin–platelet factor IV complex)
 Actually quite uncommon (0.3%–0.5%); often overdiagnosed
Actually quite uncommon (0.3%–0.5%); often overdiagnosed
 Requires recent exposure to heparin (more commonly seen with unfractionated heparin; however, can occur after exposure to the low molecular weight heparin (LMWH) as well)
Requires recent exposure to heparin (more commonly seen with unfractionated heparin; however, can occur after exposure to the low molecular weight heparin (LMWH) as well)
 Hallmark = low platelets and thrombosis
Hallmark = low platelets and thrombosis
 Discontinue heparin if the platelet count falls to <50,000 or drops >50% of the patients baseline values or if thrombosis occurs (venous>arterial, except in cardiovascular patients)
Discontinue heparin if the platelet count falls to <50,000 or drops >50% of the patients baseline values or if thrombosis occurs (venous>arterial, except in cardiovascular patients)
 Decrease in platelet count does not always precede thrombosis
Decrease in platelet count does not always precede thrombosis
 ELISA test: anti-PF4 antibodies; sensitive but specificity is low
ELISA test: anti-PF4 antibodies; sensitive but specificity is low
 Drug induced
Drug induced
 Clinical diagnosis made after fall in platelets after at least a week of exposure to a possible offending agent, with a return to normal levels after a week of discontinuing the drug
Clinical diagnosis made after fall in platelets after at least a week of exposure to a possible offending agent, with a return to normal levels after a week of discontinuing the drug
 Platelet counts < 20,000 are more commonly drug induced.
Platelet counts < 20,000 are more commonly drug induced.
 Common offenders
Common offenders
 Anticonvulsants (valproic acid, phenytoin)
Anticonvulsants (valproic acid, phenytoin)
 Cinchona alkaloids (quinine, quinidine)
Cinchona alkaloids (quinine, quinidine)
 H2 antagonists (cimetidine)
H2 antagonists (cimetidine)
 Antibiotics (vancomycin, linezolid, rifampin, sulfonamides)
Antibiotics (vancomycin, linezolid, rifampin, sulfonamides)
 Platelet inhibitors (abciximab, eptifibatide)
Platelet inhibitors (abciximab, eptifibatide)
 Antirheumatics (gold salts)
Antirheumatics (gold salts)
 Analgesics (naproxen, diclofenac, acetaminophen)
Analgesics (naproxen, diclofenac, acetaminophen)
Management and Treatment
 TTP: plasmapheresis
TTP: plasmapheresis
 Give FFP to supply metalloproteases (until plasmapheresis can be initiated)
Give FFP to supply metalloproteases (until plasmapheresis can be initiated)
 Do not give platelets (which can worsen microvascular thrombosis) unless there is severe hemorrhage or intracranial bleed.
Do not give platelets (which can worsen microvascular thrombosis) unless there is severe hemorrhage or intracranial bleed.
 Steroids have been tried for patients who have a relapse.
Steroids have been tried for patients who have a relapse.
 Other immunosuppressive agents (cyclophosphamide, cyclosporine, vincristine, rituximab) have also been used (although clinical trials guiding their use are lacking).
Other immunosuppressive agents (cyclophosphamide, cyclosporine, vincristine, rituximab) have also been used (although clinical trials guiding their use are lacking).
 HUS: supportive therapy with fluids if oliguria persists for <24 hours; can progress to requiring renal replacement therapy
HUS: supportive therapy with fluids if oliguria persists for <24 hours; can progress to requiring renal replacement therapy
 Platelet transfusions are unnecessary (as the thrombocytopenia is not often severe).
Platelet transfusions are unnecessary (as the thrombocytopenia is not often severe).
 Plasmapheresis results are equivocal.
Plasmapheresis results are equivocal.
 ITP: IV Immunoglobulin
ITP: IV Immunoglobulin
 Steroids can be used as an adjunct
Steroids can be used as an adjunct
 Splenectomy (for refractory cases).
Splenectomy (for refractory cases).
 Do not give platelets (as the “new” platelets will be subjected to the same autoimmune destruction as native platelets) unless there is severe hemorrhage or intracranial bleed.
Do not give platelets (as the “new” platelets will be subjected to the same autoimmune destruction as native platelets) unless there is severe hemorrhage or intracranial bleed.
 HIT: remove all heparin exposure (including heparin catheters and flushes)
HIT: remove all heparin exposure (including heparin catheters and flushes)
 Need to substitute with another anti-coagulating agent (Argatroban, Lepirudin)
Need to substitute with another anti-coagulating agent (Argatroban, Lepirudin)
 Do not give platelets, as they can initiate a thrombotic event.
Do not give platelets, as they can initiate a thrombotic event.
 Drug induced
Drug induced
 Remove offending agent
Remove offending agent
 Most patients do not require specific treatment; however, those with severe thrombocytopenia should receive platelet transfusions.
Most patients do not require specific treatment; however, those with severe thrombocytopenia should receive platelet transfusions.
 Can try steroids, although there is no evidence that it helps
Can try steroids, although there is no evidence that it helps
 IVIg and plasmapheresis have been used as well; however, there is no supporting evidence for their efficacy.
IVIg and plasmapheresis have been used as well; however, there is no supporting evidence for their efficacy.
SUGGESTED READINGS
Cawley MJ, Wittbrodt ET, Boyce EG, et al. Potential risk factors associated with thrombocytopenia in a surgical intensive care unit. Pharmacotherapy. 1999;19:108-113.
Hanes SD, Quarled DA, Boucher BA. Incidence and risk factors of thrombocytopenia in critically ill trauma patients. Ann Pharmacother. 1997;31:285-289.
Hui P, Cook DJ, Lim W, et al. The frequency and clinical significance of thrombocytopenia complicating critical illness: a systematic review. Chest. 2011;139:271-278.
Priziola JL, Smythe MA, Dager WE. Drug-induced thrombocytopenia in critically ill patients. Crit Care Med. 2010;38(6 Suppl):S145-S154.
Strauss R, Wehler M, Mehler K, et al. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements and outcome. Crit Care Med. 2002;30:1765-1771.
7.3
Thrombocytosis
Sheri M. Berg
Disease of increased number of platelets characterized by
 Platelet count > 450,000
Platelet count > 450,000
 Often symptomless but can predispose to thrombosis
Often symptomless but can predispose to thrombosis
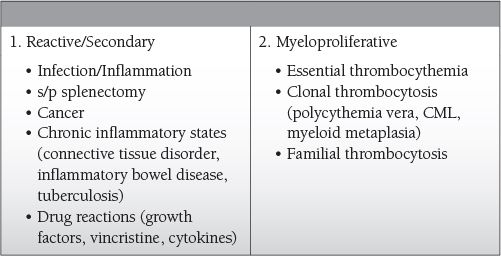
Common Causes to Remember
Epidemiology
 Most often reactive/secondary
Most often reactive/secondary
Key Pathophysiology
 Reactive thrombocytosis: increased levels of thrombopoietin, interleukin-6, cytokines (all may be produced in response to increased “stress”—inflammation, infections, and cancer)
Reactive thrombocytosis: increased levels of thrombopoietin, interleukin-6, cytokines (all may be produced in response to increased “stress”—inflammation, infections, and cancer)
 Myeloproliferative: increased levels of thrombopoietin secondary to mutations in the thrombopoietin gene
Myeloproliferative: increased levels of thrombopoietin secondary to mutations in the thrombopoietin gene
Differential Diagnosis
 Reactive/secondary thrombocytosis
Reactive/secondary thrombocytosis
 Patients will have a clinically apparent systemic disease process that can explain the elevated platelet count.
Patients will have a clinically apparent systemic disease process that can explain the elevated platelet count.
 Megakaryocytes appear normal.
Megakaryocytes appear normal.
 Platelet function is usually normal.
Platelet function is usually normal.
 Myeloproliferative
Myeloproliferative
 Patients are more likely to have complications (bleeding, thrombosis).
Patients are more likely to have complications (bleeding, thrombosis).
 Splenomegaly is more common.
Splenomegaly is more common.
 “Giant” platelets are more often seen on a peripheral blood smear.
“Giant” platelets are more often seen on a peripheral blood smear.
 Megakaryocytes appear abnormal (giant, dysplastic, increased ploidy).
Megakaryocytes appear abnormal (giant, dysplastic, increased ploidy).
 Platelet function may be abnormal.
Platelet function may be abnormal.
Management and Treatment
 Reactive/Secondary
Reactive/Secondary
 Do not usually require treatment, but the underlying disease process needs to be identified and treated accordingly.
Do not usually require treatment, but the underlying disease process needs to be identified and treated accordingly.
 Myeloproliferative
Myeloproliferative
 May require platelet lowering therapy (cytoreduction)
May require platelet lowering therapy (cytoreduction)
 If symptomatic (signs of cerebrovascular or digital ischemia), treat with hydroxyurea.
If symptomatic (signs of cerebrovascular or digital ischemia), treat with hydroxyurea.
 Anagrelide (po quinolzalone derivative) reduces platelet counts via inhibition of megakaryocyte proliferation and differentiation.
Anagrelide (po quinolzalone derivative) reduces platelet counts via inhibition of megakaryocyte proliferation and differentiation.
 Interferon alpha
Interferon alpha
 Aspirin
Aspirin
SUGGESTED READINGS
Harrison CN. Current trends in essential thrombocythemia. Br J Haematol. 2002;117:796-808.
Pearson TC. The risk of thrombosis in essential thrombocythemia and polycythemia vera. Semin Oncol. 2002;29(Suppl 10):16-21.
Shafer AI. Thrombocytosis and thrombocythemia. Blood Rev. 2001;15: 159-166.
Shafer AI. Thrombocytosis. N Engl J Med. 2004;350:2122-2129.
7.4
Neutropenia
Sheri M. Berg
Definition
 Disease of decreased number of neutrophils characterized by
Disease of decreased number of neutrophils characterized by
 Absolute neutrophil count (ANC) < 1,500/mm3
Absolute neutrophil count (ANC) < 1,500/mm3
 Classified as mild, moderate, or severe
Classified as mild, moderate, or severe
 Risk of infection increases as ANC decreases
Risk of infection increases as ANC decreases
 Common features include fevers and infections
Common features include fevers and infections
 Severe neutropenia and increased risk of severe infections occur when the ANC < 500/mm3.
Severe neutropenia and increased risk of severe infections occur when the ANC < 500/mm3.
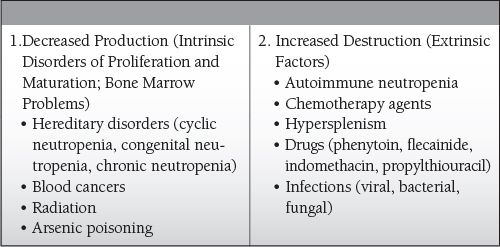
Common Causes to Remember
Epidemiology
 Females > males
Females > males
 Elderly > young
Elderly > young
 African Americans > whites
African Americans > whites
Key Pathophysiology
 Neutrophils are an imperative component of the host defense system.
Neutrophils are an imperative component of the host defense system.
 Oral mucosal ulceration and gingivitis are common problems, as bacteria overwhelm the oral pharynx in those who are neutropenic.
Oral mucosal ulceration and gingivitis are common problems, as bacteria overwhelm the oral pharynx in those who are neutropenic.
 Neutropenia secondary to chemotherapy drugs tends to result in the most severe infections.
Neutropenia secondary to chemotherapy drugs tends to result in the most severe infections.
 Infections can be severe, necessitating intensive care unit (ICU) admission.
Infections can be severe, necessitating intensive care unit (ICU) admission.
 Usually secondary to “neutropenic fever”
Usually secondary to “neutropenic fever”
 Oral pharynx and gastrointestinal tract can be severely affected.
Oral pharynx and gastrointestinal tract can be severely affected.
 Chronic neutropenics tend to have functional monocytes and intact immune function.
Chronic neutropenics tend to have functional monocytes and intact immune function.
 Infections tend to be less severe as compared to those with neutropenia caused by chemotherapy.
Infections tend to be less severe as compared to those with neutropenia caused by chemotherapy.
 It is important to note that normal, systemic signs of infection may be blunted in neutropenics, but they will present with fever, possibly as their only marker of inflammation and infection.
It is important to note that normal, systemic signs of infection may be blunted in neutropenics, but they will present with fever, possibly as their only marker of inflammation and infection.
Differential Diagnosis
 Any neutropenic patient must be thoroughly worked up, as they often simply present with a fever.
Any neutropenic patient must be thoroughly worked up, as they often simply present with a fever.
 Fever of unknown origin
Fever of unknown origin
 Drug induced
Drug induced
 Infection: bacterial, viral, fungal
Infection: bacterial, viral, fungal
 CBC with differential and evaluation of a peripheral blood smear
CBC with differential and evaluation of a peripheral blood smear
 Shock and respiratory failure are the leading causes for neutropenic admissions to the ICU.
Shock and respiratory failure are the leading causes for neutropenic admissions to the ICU.
 Macrophage activation syndrome (aka lymphohistiocytic activation syndrome)
Macrophage activation syndrome (aka lymphohistiocytic activation syndrome)
 Can be seen in a patient who is neutropenic
Can be seen in a patient who is neutropenic
 Can cause neutropenia
Can cause neutropenia
 Vasoplegic shock and multisystem organ failure can ensue
Vasoplegic shock and multisystem organ failure can ensue
 Three key features are fever, hepatosplenomegaly, and thrombocytopenia
Three key features are fever, hepatosplenomegaly, and thrombocytopenia
 Treated with steroids and etoposide (chemotherapy drug)
Treated with steroids and etoposide (chemotherapy drug)
Management and Treatment
 If a specific drug is the assumed offending agent, remove it immediately.
If a specific drug is the assumed offending agent, remove it immediately.
 If a neutropenic patient presents with a fever, antibiotic therapy should be initiated and provide coverage against
If a neutropenic patient presents with a fever, antibiotic therapy should be initiated and provide coverage against
 Gram-positive cocci (staph and strep)
Gram-positive cocci (staph and strep)
 Gram-negative rods (pseudomonas)
Gram-negative rods (pseudomonas)
 Antifungals (if a patient has not improved within a week of what should be adequate antibacterial coverage, antifungal coverage should be added)
Antifungals (if a patient has not improved within a week of what should be adequate antibacterial coverage, antifungal coverage should be added)
 Hematopoietic growth factors
Hematopoietic growth factors Granulocyte colony-stimulating factor—increases neutrophil count and function
Granulocyte colony-stimulating factor—increases neutrophil count and function
 Isolation rooms
Isolation rooms
SUGGESTED READINGS
Bhatt V, Saleem A. Review: drug induced neutropenia- pathophysiology, clinical features, and management. Ann Clin Lab Sci. 2004;34(2):131-137.
Boxer L, Dale DC. Neutropenia: causes and consequences. Semin Hematol. 2002;39(2):75-81.
Hsieh MM, Everhart JE, Byrd-Holt DD, et al. Prevalence of neutropenia in the US population: age, sex, smoking status, and ethnic differences. Ann Intern Med. 2007;146(7):486-492.
Hughes WT, Armstrong D, Bodey GP, et al. 2002 guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clin Infect Dis. 2002;34:730-751.
7.5
Hemoglobinopathies
Jarone Lee
Introduction
 Hemoglobinopathies include a group of disorders of globin chain synthesis, which include the sickle cell syndromes, thalassemias, sickle cell and thalassemia combinations, unstable hemoglobins, and toxic exposures, such as methemoglobinemia.
Hemoglobinopathies include a group of disorders of globin chain synthesis, which include the sickle cell syndromes, thalassemias, sickle cell and thalassemia combinations, unstable hemoglobins, and toxic exposures, such as methemoglobinemia.
 The theory is that the hereditary forms developed over time as a way for the body to resist blood-born infectious diseases, mostly malaria.
The theory is that the hereditary forms developed over time as a way for the body to resist blood-born infectious diseases, mostly malaria.
Epidemiology
 Hereditary hemoglobinopathies are clustered in areas where malaria is endemic, affecting around 200 million individuals worldwide.
Hereditary hemoglobinopathies are clustered in areas where malaria is endemic, affecting around 200 million individuals worldwide.
 8% of American blacks are heterozygous for sickle cell disease
8% of American blacks are heterozygous for sickle cell disease
 1 in 400 American blacks are homozygous for sickle cell disease.
1 in 400 American blacks are homozygous for sickle cell disease.
 Acute Chest Syndrome
Acute Chest Syndrome
 Highest in homozygous sickle cell disease
Highest in homozygous sickle cell disease
 2 to 4 years of age: 25.3 per 100 patient-years
2 to 4 years of age: 25.3 per 100 patient-years
 > 20 years of age: 8.8 per 100 patient-years
> 20 years of age: 8.8 per 100 patient-years
 15% of American blacks are alpha-thalassemia carriers.
15% of American blacks are alpha-thalassemia carriers.
 10%–15% of people from the Mediterranean and Southeast Asia have beta-thalassemia.
10%–15% of people from the Mediterranean and Southeast Asia have beta-thalassemia.

Key Pathophysiology
 Hemoglobin is made of globin polypeptide chains arranged in a tetramer.
Hemoglobin is made of globin polypeptide chains arranged in a tetramer.
 The tetramer typically divided into two alpha chains and two beta chains.
The tetramer typically divided into two alpha chains and two beta chains.
 Each hemoglobin polypeptide chain interacts with one heme moiety and an iron atom in the ferrous state.
Each hemoglobin polypeptide chain interacts with one heme moiety and an iron atom in the ferrous state.
 Each heme-ferrous complex can bind one oxygen molecule.
Each heme-ferrous complex can bind one oxygen molecule.
 Therefore, each hemoglobin tetramer can bind four oxygen molecules.
Therefore, each hemoglobin tetramer can bind four oxygen molecules.
 Mutations in the amino acid sequences of the alpha or beta chains can cause disease and clinical symptoms.
Mutations in the amino acid sequences of the alpha or beta chains can cause disease and clinical symptoms.
 The alpha chain is 141 amino acids encoded on chromosome 16, and the beta chain is 146 amino acids encoded on chromosome 11.
The alpha chain is 141 amino acids encoded on chromosome 16, and the beta chain is 146 amino acids encoded on chromosome 11.
 Known areas of mutations that cause clinical symptoms include: (1) a key histidine in the F helix; (2) connections between the Alpha1 and Beta 1 subunits; and (3) connections between the Alpha1 and Beta2 subunits.
Known areas of mutations that cause clinical symptoms include: (1) a key histidine in the F helix; (2) connections between the Alpha1 and Beta 1 subunits; and (3) connections between the Alpha1 and Beta2 subunits.
 Types of hemoglobin
Types of hemoglobin
 During the first trimester, the fetus has primarily embryonic hemoglobins: Gower 1; Gower 2; and Portland 1.
During the first trimester, the fetus has primarily embryonic hemoglobins: Gower 1; Gower 2; and Portland 1.
 By 34 to 36 week gestation, Fetal hemoglobin (Hb F) comprises 90% to 95%, with the rest represented by adult hemoglobin (Hb A).
By 34 to 36 week gestation, Fetal hemoglobin (Hb F) comprises 90% to 95%, with the rest represented by adult hemoglobin (Hb A).
 At term, Hb F comprises 53% to 95%, with Hb A comprising around 20% to 30% of total hemoglobin.
At term, Hb F comprises 53% to 95%, with Hb A comprising around 20% to 30% of total hemoglobin.
 Persistently elevated Hb F is seen in hemoglobinopathies, but can also be seen in the following infants:
Persistently elevated Hb F is seen in hemoglobinopathies, but can also be seen in the following infants:
 Small for gestational age
Small for gestational age
 Chronic hypoxia
Chronic hypoxia
 Trisomy 13
Trisomy 13
 Hb F levels stays elevated during the neonatal period, and decreases to <3% by 6 months of life.
Hb F levels stays elevated during the neonatal period, and decreases to <3% by 6 months of life.
 At birth, the infant starts producing hemoglobin A2 (Hb A2) and reaches adult levels by 6 months.
At birth, the infant starts producing hemoglobin A2 (Hb A2) and reaches adult levels by 6 months.
 Because of the predominance of alpha chain hemoglobins in infancy, typically alpha chain hemoglobinopathies present in the neonatal period, whereas beta chain abnormalities present after 3 months of age.
Because of the predominance of alpha chain hemoglobins in infancy, typically alpha chain hemoglobinopathies present in the neonatal period, whereas beta chain abnormalities present after 3 months of age.
 Common pathway for hereditary hemoglobinopathies
Common pathway for hereditary hemoglobinopathies
 Chromosomal mutations in the amino acids associated with the alpha or beta chains will produce clinical syndromes if they alter solubility and/or oxygen-binding affinity.
Chromosomal mutations in the amino acids associated with the alpha or beta chains will produce clinical syndromes if they alter solubility and/or oxygen-binding affinity.
 This leads to an inadequate supply of hemoglobin, which leads to the destruction of red cells and associated precursors.
This leads to an inadequate supply of hemoglobin, which leads to the destruction of red cells and associated precursors.
 Depending on the number and location of mutations, clinical severity can range from inutero death (e.g., hydropsfetalis) to asymptomatic carriers with mild anemia.
Depending on the number and location of mutations, clinical severity can range from inutero death (e.g., hydropsfetalis) to asymptomatic carriers with mild anemia.
 Specific hereditary hemoglobinopathies
Specific hereditary hemoglobinopathies
 Sickle Cell Syndromes: caused by mutation in beta-chain that changes the sixth amino acid from glutamic acid to valine (Hb S)
Sickle Cell Syndromes: caused by mutation in beta-chain that changes the sixth amino acid from glutamic acid to valine (Hb S)
 When deoxygenated, Hb S will polymerize, which leads to red-cell hardening and the characteristic ‘sickle cell’.
When deoxygenated, Hb S will polymerize, which leads to red-cell hardening and the characteristic ‘sickle cell’.
 Occluded capillary vessels from the polymers leads to microinfarction of organs and pain.
Occluded capillary vessels from the polymers leads to microinfarction of organs and pain.
 Furthermore, hemolysis occurs as the body attempts to digest the polymers.
Furthermore, hemolysis occurs as the body attempts to digest the polymers.
 Unstable Hemoglobins: mutations that decrease solubility, typically form inclusion bodies in red cells called Heinz bodies
Unstable Hemoglobins: mutations that decrease solubility, typically form inclusion bodies in red cells called Heinz bodies
 The spleen removes these Heinz bodies, leaving defective red blood cells, which are subsequently destroyed by the body at a rapid rate.
The spleen removes these Heinz bodies, leaving defective red blood cells, which are subsequently destroyed by the body at a rapid rate.
 As a result, even patients that are heterozygous are symptomatic.
As a result, even patients that are heterozygous are symptomatic.
 Thalassemia: inherited disorder of alpha and beta subunits of hemoglobin
Thalassemia: inherited disorder of alpha and beta subunits of hemoglobin
 A reduced supply of the affect hemoglobin will cause decreased production and accumulation of the other subunits.
A reduced supply of the affect hemoglobin will cause decreased production and accumulation of the other subunits.
 Heterozygous individuals typically only manifest with mild anemia.
Heterozygous individuals typically only manifest with mild anemia.
 Homozygous individuals typically are more severe, since the red cell precursors are destroyed in the bone marrow, with the few red cells that survive ultimately destroyed by the spleen.
Homozygous individuals typically are more severe, since the red cell precursors are destroyed in the bone marrow, with the few red cells that survive ultimately destroyed by the spleen.
 This leads to a profound hemolytic anemia.
This leads to a profound hemolytic anemia.
 The body adapts by hypertrophy of the bone marrow.
The body adapts by hypertrophy of the bone marrow.
 This causes characteristic bone abnormalities in patients, such as ‘chipmunk’ facies.
This causes characteristic bone abnormalities in patients, such as ‘chipmunk’ facies.
 Acquired hemoglobinopathies
Acquired hemoglobinopathies
 Patients with acquired hemoglobinopathies typically have normal genetic hemoglobin structures but are either poisoned by toxins (e.g., methemoglobinemia) or have a myeloproliferative disorder.
Patients with acquired hemoglobinopathies typically have normal genetic hemoglobin structures but are either poisoned by toxins (e.g., methemoglobinemia) or have a myeloproliferative disorder.
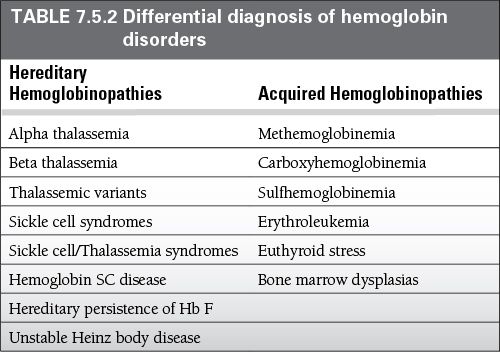
Differential Diagnosis
 The differential diagnosis for anemia is large; however, consider testing for hemoglobinopathies in patients with the following:
The differential diagnosis for anemia is large; however, consider testing for hemoglobinopathies in patients with the following:
 History of hydrops fetalis during pregnancy
History of hydrops fetalis during pregnancy
 Unexplained anemia and splenomegaly
Unexplained anemia and splenomegaly
 Neonatal or infantile anemia with Hb F
Neonatal or infantile anemia with Hb F
 Neonatal of infantile anemia with low Hb A
Neonatal of infantile anemia with low Hb A
 Unexplained microcytosis
Unexplained microcytosis
 Clinical features suggestive of sickle cell
Clinical features suggestive of sickle cell
 Unexplained hemolysis
Unexplained hemolysis
 Unexplained irregularly contracted cells
Unexplained irregularly contracted cells
 Unexplained polycythemia
Unexplained polycythemia
 Unexplained cyanosis with normal oxygen saturations
Unexplained cyanosis with normal oxygen saturations
Diagnosis
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


