Abdominal perfusion pressure = Mean Arterial Pressure – Intraabdominal Pressure (APP = MAP – IAP)
 ACS refers to multiple organ dysfunction due to increased intraabdominal pressure, typically >20 to 25 mmHg.
ACS refers to multiple organ dysfunction due to increased intraabdominal pressure, typically >20 to 25 mmHg.
 Abdominal hypertension refers to acute or chronic elevation of the intraabdominal pressure, typically >12 mmHg (normal abdominal pressure is around 5 mmHg).
Abdominal hypertension refers to acute or chronic elevation of the intraabdominal pressure, typically >12 mmHg (normal abdominal pressure is around 5 mmHg).
Common Causes to Remember
 Massive resuscitation and transfusions leading to bowel wall edema
Massive resuscitation and transfusions leading to bowel wall edema
 Intraperitoneal or retroperitoneal hemorrhage
Intraperitoneal or retroperitoneal hemorrhage
 Bowel distention due to obstruction, ileus, or acute inflammation; bowel reperfusion injury from ischemia of any cause
Bowel distention due to obstruction, ileus, or acute inflammation; bowel reperfusion injury from ischemia of any cause
 Ascites/any space-occupying intraabdominal process, mass, or fluid
Ascites/any space-occupying intraabdominal process, mass, or fluid
 Loss of domain/abdominal closure under tension, usually after repair of complex ventral hernias
Loss of domain/abdominal closure under tension, usually after repair of complex ventral hernias
Types of ACS
 Primary: primary etiology lies within the abdomen (e.g., excessive bowel wall edema or distention)
Primary: primary etiology lies within the abdomen (e.g., excessive bowel wall edema or distention)
 Secondary: from external causes (e.g., circumferential burn eschar)
Secondary: from external causes (e.g., circumferential burn eschar)
 Acute: e.g., acute bowel wall edema from massive resuscitation
Acute: e.g., acute bowel wall edema from massive resuscitation
 Chronic: e.g., late stages of cirrhosis with massive ascites
Chronic: e.g., late stages of cirrhosis with massive ascites
Epidemiology
 Incidence of intraabdominal hypertension in medical ICUs is 8%.
Incidence of intraabdominal hypertension in medical ICUs is 8%.
 Incidence of true compartment syndrome in trauma patients is 2% to 9%.
Incidence of true compartment syndrome in trauma patients is 2% to 9%.
Key Pathophysiology
 Upward displacement of the diaphragm results in decreased thoracic volumes and pulmonary compliance, which lead to an increase in peak airway pressures, ventilation and perfusion mismatch, hypoxia, hypercapnia, and acidosis.
Upward displacement of the diaphragm results in decreased thoracic volumes and pulmonary compliance, which lead to an increase in peak airway pressures, ventilation and perfusion mismatch, hypoxia, hypercapnia, and acidosis.
 Compression of the inferior vena cava results in decreased cardiac venous return and increased peripheral vascular resistance. (As a result, blood pressure, cardiac output, cardiac index, and right atrial and pulmonary artery occlusion pressures decrease. Systemic delivery and consumption of O2 decreases.)
Compression of the inferior vena cava results in decreased cardiac venous return and increased peripheral vascular resistance. (As a result, blood pressure, cardiac output, cardiac index, and right atrial and pulmonary artery occlusion pressures decrease. Systemic delivery and consumption of O2 decreases.)
 Direct compression of the kidneys and obstruction of venous outflow result in a decrease in the glomerular filtration rate and urine output.
Direct compression of the kidneys and obstruction of venous outflow result in a decrease in the glomerular filtration rate and urine output.
 Compression of the mesenteric vasculature leads to a decrease in splanchnic perfusion, mesenteric venous hypertension, intestinal edema, and visceral edema.
Compression of the mesenteric vasculature leads to a decrease in splanchnic perfusion, mesenteric venous hypertension, intestinal edema, and visceral edema.
 Elevated central venous pressure interferes with cerebral venous outflow and results in increased intracranial pressure.
Elevated central venous pressure interferes with cerebral venous outflow and results in increased intracranial pressure.
 Previous pregnancy, cirrhosis, morbid obesity can chronically increase abdominal wall compliance and may be protective.
Previous pregnancy, cirrhosis, morbid obesity can chronically increase abdominal wall compliance and may be protective.
Clinical Manifestations
 Distended, tense abdomen
Distended, tense abdomen
 Hypoxia, hypercapnia, and high peak inspiratory pressures with decreased compliance in the mechanically ventilated patients
Hypoxia, hypercapnia, and high peak inspiratory pressures with decreased compliance in the mechanically ventilated patients
 Shortness of breath in extubated patients
Shortness of breath in extubated patients
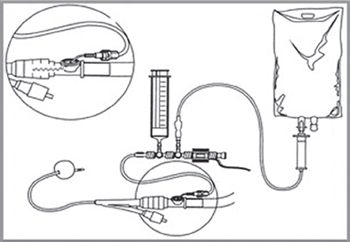
Figure 4.1.1 Repeat frequently (every 4 to 6 hours). Monitor the trend.
 Elevated central venous and pulmonary capillary wedge pressures
Elevated central venous and pulmonary capillary wedge pressures
 Oliguria and eventually anuria
Oliguria and eventually anuria
 Decreased cardiac output, hypotension, acidosis, vasopressor requirement
Decreased cardiac output, hypotension, acidosis, vasopressor requirement
 Decreased intestinal, renal, hepatic blood flow
Decreased intestinal, renal, hepatic blood flow
 Signs of increased intracranial pressure
Signs of increased intracranial pressure
Diagnosis/Measurement of Intraabdominal Pressure
 Bladder pressure is an indirect measure of intraperitoneal pressure; 50 mL of sterile saline is instilled into the bladder via a Foley catheter with the drainage tube clamped, while the aspiration port is attached to a pressure transducer (zeroed at the level of the pubic symphysis, Fig. 4.1.1).
Bladder pressure is an indirect measure of intraperitoneal pressure; 50 mL of sterile saline is instilled into the bladder via a Foley catheter with the drainage tube clamped, while the aspiration port is attached to a pressure transducer (zeroed at the level of the pubic symphysis, Fig. 4.1.1).
Management and Treatment
 Surgical decompression is typically definitive! Abdomen is left open (usually with a negative pressure Vac dressing), until the etiology of the ACS improves.
Surgical decompression is typically definitive! Abdomen is left open (usually with a negative pressure Vac dressing), until the etiology of the ACS improves.
 Paralytics and diuretics may have a role in the management of intraabdominal hypertension.
Paralytics and diuretics may have a role in the management of intraabdominal hypertension.
 Paracentesis is done for ascites and escharotomy for burn eschars.
Paracentesis is done for ascites and escharotomy for burn eschars.
SUGGESTED READINGS
Joynt GM, Wai JK. Intra-abdominal hypertension and abdominal compartment syndrome—making progress? Anaesth Intensive Care. 2012;40(1):11-13.
Khan S, Verma AK, Ahmad SM, et al. Analyzing intra-abdominal pressures and outcomes in patients undergoing emergency laparotomy. J Emerg Trauma Shock. 2010;3(4):318-325.
Malbrain ML, Cheatham ML. Definitions and pathophysiological implications of intra-abdominal hypertension and abdominal compartment syndrome. Am Surg. 2011;77(Suppl 1):S6-S11.
Malbrain ML, Chiumello D, Pelosi P, et al. Incidence and prognosis of intraabdominal hypertension in a mixed population of critically ill patients: a multiple-center epidemiological study. Crit Care Med. 2005;33(2):315-322.
Raeburn CD, Moore EE, Biffl WL, et al. The abdominal compartment syndrome is a morbid complication of postinjury damage control surgery. Am J Surg. 2001;182(6):542-546.
4.2
Acute Pancreatitis
Antonios C. Sideris and Peter J. Fagenholz
Introduction
 Inflammatory disease of the pancreas. Categories:
Inflammatory disease of the pancreas. Categories:
 Mild (edematous, interstitial) acute pancreatitis
Mild (edematous, interstitial) acute pancreatitis
 Severe acute pancreatitis (SAP): one or more of the following:
Severe acute pancreatitis (SAP): one or more of the following:
 Failure of one or more organ systems (respiratory, renal, gastrointestinal, circulatory, disseminated intravascular coagulation (DIC)) at any time during the course of the disease
Failure of one or more organ systems (respiratory, renal, gastrointestinal, circulatory, disseminated intravascular coagulation (DIC)) at any time during the course of the disease
 Three or more Ranson’s criteria (see “severity scoring”) or Apache II of eight or above
Three or more Ranson’s criteria (see “severity scoring”) or Apache II of eight or above
 Local complications: necrosis, pseudocyst, and/or abscess
Local complications: necrosis, pseudocyst, and/or abscess
 Moderate pancreatitis: SAP without organ failure
Moderate pancreatitis: SAP without organ failure
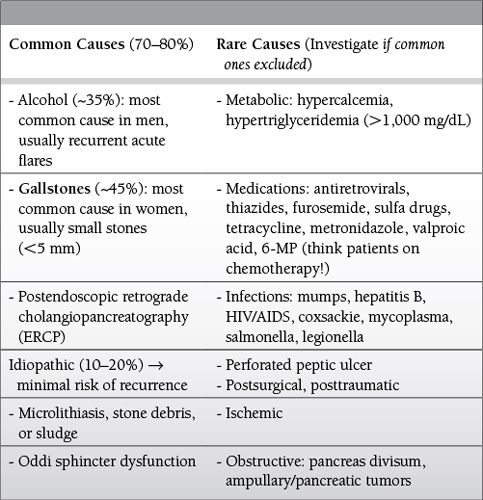
Common Causes to Remember
Epidemiology
 Increasing incidence, ranging from 5 to 44 cases per 100,000 population
Increasing incidence, ranging from 5 to 44 cases per 100,000 population
 Severe pancreatitis incidence has remained stable over time (10% to 20% of all cases).
Severe pancreatitis incidence has remained stable over time (10% to 20% of all cases).
 Slight increase with age, blacks more than whites, females more than males
Slight increase with age, blacks more than whites, females more than males
 Only 3% to 7% of patients with cholelithiasis and 10% of alcoholics develop acute pancreatitis.
Only 3% to 7% of patients with cholelithiasis and 10% of alcoholics develop acute pancreatitis.
 Up to 75% have elevated amylase post-ERCP, but only 5% develop overt clinical pancreatitis.
Up to 75% have elevated amylase post-ERCP, but only 5% develop overt clinical pancreatitis.
Key Pathophysiology
 Unregulated protease activation within pancreatic acinar cells, instead of inside the small bowel lumen
Unregulated protease activation within pancreatic acinar cells, instead of inside the small bowel lumen
 Protective mechanisms (enzyme compartmentalization, specific trypsin inhibitors such as serine protease inhibitor Kazal type 1 (SPINK1), low intracellular Ca++) are overwhelmed.
Protective mechanisms (enzyme compartmentalization, specific trypsin inhibitors such as serine protease inhibitor Kazal type 1 (SPINK1), low intracellular Ca++) are overwhelmed.
 Trypsinogen converted to trypsin by hydrolysis of N-terminal trypsinogen activating peptide (TAP), leading to activation of pancreatic proteases (phospholipase A2, chymotrypsin, elastase), complement, kallikrein-kinin, proinflammatory interleukins (TNFa, IL-1, IL-6, IL-8), coagulation, and fibrinolysis.
Trypsinogen converted to trypsin by hydrolysis of N-terminal trypsinogen activating peptide (TAP), leading to activation of pancreatic proteases (phospholipase A2, chymotrypsin, elastase), complement, kallikrein-kinin, proinflammatory interleukins (TNFa, IL-1, IL-6, IL-8), coagulation, and fibrinolysis.
 Autodigestion of the gland (with or without SIRS)
Autodigestion of the gland (with or without SIRS)
 Mechanisms of trypsin activation depend on etiology, largely debated.
Mechanisms of trypsin activation depend on etiology, largely debated.
 Mechanical: gallstone, ERCP, trauma, pancreas divisum
Mechanical: gallstone, ERCP, trauma, pancreas divisum
 Obstruction of the ampulla by the stone, tumor or edema by the stone passage, and decreased flow from secondary pancreatic duct (Santorini) in pancreas divisum leading to increased intrapancreatic duct pressures and/or
Obstruction of the ampulla by the stone, tumor or edema by the stone passage, and decreased flow from secondary pancreatic duct (Santorini) in pancreas divisum leading to increased intrapancreatic duct pressures and/or
 Reflux of bile into pancreatic duct during transient ampulla obstruction
Reflux of bile into pancreatic duct during transient ampulla obstruction
 Systemic: EtOH, drugs, hypertriglyceridemia, hyper Ca++, infections, and toxins: exact mechanisms remain unclear.
Systemic: EtOH, drugs, hypertriglyceridemia, hyper Ca++, infections, and toxins: exact mechanisms remain unclear.
Diagnosis
 Combination of clinical, laboratory, and imaging findings
Combination of clinical, laboratory, and imaging findings
 Clinical
Clinical
 Epigastric pain, sometimes radiating to the back or shoulder, accompanied by nausea and/or vomiting is the most common clinical presentation.
Epigastric pain, sometimes radiating to the back or shoulder, accompanied by nausea and/or vomiting is the most common clinical presentation.
 Signs of shock in fulminant attacks
Signs of shock in fulminant attacks
 Tenderness on palpation +/– guarding
Tenderness on palpation +/– guarding
 Jaundice if there is concurrent biliary obstruction
Jaundice if there is concurrent biliary obstruction
 Palpable abdominal mass if a pancreatic collection or pseudocyst is present
Palpable abdominal mass if a pancreatic collection or pseudocyst is present
 Ecchymosis of the periumbilical (Cullen sign) and/or flank area (Grey-Turner sign) are rare and nonspecific (retroperitoneal hemorrhage).
Ecchymosis of the periumbilical (Cullen sign) and/or flank area (Grey-Turner sign) are rare and nonspecific (retroperitoneal hemorrhage).
 Laboratory
Laboratory
 Serum amylase and lipase more than three times the upper limit of normal is sensitive and specific for diagnosis of AP if clinical signs are present.
Serum amylase and lipase more than three times the upper limit of normal is sensitive and specific for diagnosis of AP if clinical signs are present.
 Serum amylase rises in 6 to 12 hours, remains elevated for 3 to 5 days. Level does not predict severity. Urine amylase offers no benefit over serum measurement.
Serum amylase rises in 6 to 12 hours, remains elevated for 3 to 5 days. Level does not predict severity. Urine amylase offers no benefit over serum measurement.
 Serum lipase is potentially more specific than serum amylase: useful in patients with delayed presentation, may remain elevated for up to 2 weeks.
Serum lipase is potentially more specific than serum amylase: useful in patients with delayed presentation, may remain elevated for up to 2 weeks.
 ALT more than three times the upper limit of normal may be specific for gallstone pancreatitis.
ALT more than three times the upper limit of normal may be specific for gallstone pancreatitis.
 Imaging findings
Imaging findings
 Imaging is not necessary to make the diagnosis if the clinical picture and serum tests are consistent with acute pancreatitis.
Imaging is not necessary to make the diagnosis if the clinical picture and serum tests are consistent with acute pancreatitis.
 Abdominal CT with intravenous contrast
Abdominal CT with intravenous contrast
 Gold standard imaging modality, but not necessary at presentation
Gold standard imaging modality, but not necessary at presentation
 Most useful at presentation to identify potential alternative diagnoses
Most useful at presentation to identify potential alternative diagnoses
 Useful later in the course of disease to identify necrosis
Useful later in the course of disease to identify necrosis
 Abdominal ultrasound
Abdominal ultrasound
 Best modality to identify cholelithiasis, common bile duct dilatation
Best modality to identify cholelithiasis, common bile duct dilatation
 Gallstones confirm etiology of gallstone pancreatitis in the absence of a more apparent cause. If seen, pancreas may appear edematous.
Gallstones confirm etiology of gallstone pancreatitis in the absence of a more apparent cause. If seen, pancreas may appear edematous.
 Endoscopic ultrasound is the most accurate modality for choledocholithiasis, but is invasive—rarely necessary.
Endoscopic ultrasound is the most accurate modality for choledocholithiasis, but is invasive—rarely necessary.
Severity Scoring
 No system is superior.
No system is superior.
 C-reactive protein ≥ 150 mg/L at 48 hours distinguishes mild versus severe attack.
C-reactive protein ≥ 150 mg/L at 48 hours distinguishes mild versus severe attack.
 Procalcitonin: similar to Apache II and Ranson criteria, cutoffs remain to be determined
Procalcitonin: similar to Apache II and Ranson criteria, cutoffs remain to be determined
 Apache II: useful index to classify the severity of the disease, but cumbersome to use.
Apache II: useful index to classify the severity of the disease, but cumbersome to use.
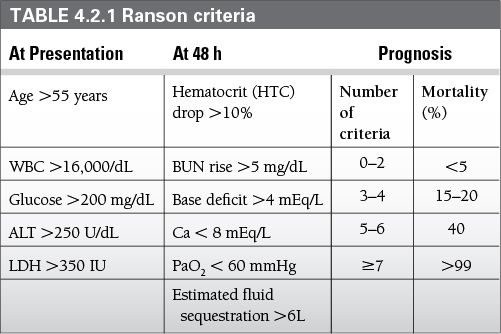
 Ranson criteria: require 48 hours of clinical information to calculate (Table 4.2.1)
Ranson criteria: require 48 hours of clinical information to calculate (Table 4.2.1)
 CT severity index: estimates the degree of necrosis and predicts higher morbidity and mortality. IV contrast administration is necessary (Table 4.2.2).
CT severity index: estimates the degree of necrosis and predicts higher morbidity and mortality. IV contrast administration is necessary (Table 4.2.2).
 Bedside index of severity in acute pancreatitis (BISAP)
Bedside index of severity in acute pancreatitis (BISAP)
 BUN ≥ 25 mg/dL
BUN ≥ 25 mg/dL
 Impaired mental status
Impaired mental status
 SIRS
SIRS
 Age > 60 years
Age > 60 years
 Pleural effusion
Pleural effusion
 Similar predictive ability for mortality as Apache II
Similar predictive ability for mortality as Apache II
 Harmless acute pancreatitis score (HAPS)
Harmless acute pancreatitis score (HAPS)
 Rebound tenderness/guarding
Rebound tenderness/guarding

 Creatinine ≥ 2 mg/dL
Creatinine ≥ 2 mg/dL
 Hematocrit >43% in men and >39.6% in women
Hematocrit >43% in men and >39.6% in women
 Can be calculated within 30 minutes of admission
Can be calculated within 30 minutes of admission
 98% predictive ability of patients with a benign course if all three parameters are negative
98% predictive ability of patients with a benign course if all three parameters are negative
Differential Diagnosis
 Other causes of epigastric/right upper quadrant abdominal pain: acute cholecystitis, perforated viscus, peptic ulcer, mesenteric ischemia, small bowel obstruction, ruptured abdominal aortic aneurysm (AAA), acute myocardial infarction, MI, pneumonia
Other causes of epigastric/right upper quadrant abdominal pain: acute cholecystitis, perforated viscus, peptic ulcer, mesenteric ischemia, small bowel obstruction, ruptured abdominal aortic aneurysm (AAA), acute myocardial infarction, MI, pneumonia
 Causes of hyperamylasemia: salivary tumor/inflammation, bowel obstruction, mesenteric ischemia, acute cholecystitis, ovarian tumor, ectopic pregnancy, macroamylasemia, renal failure, anorexia nervosa, liver disease
Causes of hyperamylasemia: salivary tumor/inflammation, bowel obstruction, mesenteric ischemia, acute cholecystitis, ovarian tumor, ectopic pregnancy, macroamylasemia, renal failure, anorexia nervosa, liver disease
 Always assess hyperamylasemia in conjunction with the clinical picture.
Always assess hyperamylasemia in conjunction with the clinical picture.
 False negative amylase levels: hypertriglyceridemia, recurrent attacks of alcoholic pancreatitis, resulting in “burned out pancreas”
False negative amylase levels: hypertriglyceridemia, recurrent attacks of alcoholic pancreatitis, resulting in “burned out pancreas”
Management and Treatment
 Fluid resuscitation
Fluid resuscitation
 Patients with SAP need ICU level monitoring for management of end-organ dysfunction.
Patients with SAP need ICU level monitoring for management of end-organ dysfunction.
 Fluid resuscitation is critical. The exact rate remains controversial, with renal dysfunction and systemic hypoperfusion complicating underresuscitation and ARDS and abdominal compartment syndrome complicating excessive fluid administration. Lactated Ringer’s has been shown to be superior to normal saline by reducing inflammatory mediator production—avoid in hypercalcemia-induced pancreatitis because it contains 3 mEq/L calcium.
Fluid resuscitation is critical. The exact rate remains controversial, with renal dysfunction and systemic hypoperfusion complicating underresuscitation and ARDS and abdominal compartment syndrome complicating excessive fluid administration. Lactated Ringer’s has been shown to be superior to normal saline by reducing inflammatory mediator production—avoid in hypercalcemia-induced pancreatitis because it contains 3 mEq/L calcium.
 Pain control
Pain control
 Intravenous opiates are standard.
Intravenous opiates are standard.
 Morphine has not been shown to cause or exacerbate AP attacks.
Morphine has not been shown to cause or exacerbate AP attacks.
 Nutritional support
Nutritional support
 Mild attacks: initially NPO, progress to oral feedings when the pain and nausea subside. Advance from clear liquids to solid food as tolerated
Mild attacks: initially NPO, progress to oral feedings when the pain and nausea subside. Advance from clear liquids to solid food as tolerated
 Alternatively, a low-fat solid diet has been shown recently to be as safe and may result in improved outcomes.
Alternatively, a low-fat solid diet has been shown recently to be as safe and may result in improved outcomes.
 Patients not expected to resume oral feedings for 5 to 7 days (e.g., severe attacks) should receive nutritional support.
Patients not expected to resume oral feedings for 5 to 7 days (e.g., severe attacks) should receive nutritional support.
 Enteral feeding is the preferred mode of nutritional support, as it prevents atrophy of the intestinal mucosa and bacterial translocation, thus leading to improved mortality, lower incidence of organ failure, systemic infection, need for surgery, and potentially a shorter hospital stay.
Enteral feeding is the preferred mode of nutritional support, as it prevents atrophy of the intestinal mucosa and bacterial translocation, thus leading to improved mortality, lower incidence of organ failure, systemic infection, need for surgery, and potentially a shorter hospital stay.
 Oral or nasogastric feeding can be tried first, with jejunal feeding reserved for patients who cannot tolerate these approaches.
Oral or nasogastric feeding can be tried first, with jejunal feeding reserved for patients who cannot tolerate these approaches.
 Total parenteral nutrition (TPN): use only in patients who cannot tolerate enteral feeding.
Total parenteral nutrition (TPN): use only in patients who cannot tolerate enteral feeding.
 Avoid administration as an adjunct to enteral feedings, as it has been associated with a higher mortality, increased infectious complications, and longer ICU and hospital length of stay.
Avoid administration as an adjunct to enteral feedings, as it has been associated with a higher mortality, increased infectious complications, and longer ICU and hospital length of stay.
 Antibiotic prophylaxis
Antibiotic prophylaxis
 This has no role in mild pancreatitis.
This has no role in mild pancreatitis.
 Prophylactic administration for noninfected necrosis remains highly controversial.
Prophylactic administration for noninfected necrosis remains highly controversial.
 A recent Cochrane metaanalysis failed to show a mortality benefit or a decrease in infectious complications.
A recent Cochrane metaanalysis failed to show a mortality benefit or a decrease in infectious complications.
 At present, prophylactic administration of antibiotics in noninfected necrosis is not advisable.
At present, prophylactic administration of antibiotics in noninfected necrosis is not advisable.
 Prophylactic probiotic administration is discouraged, as it has been associated with bowel ischemia.
Prophylactic probiotic administration is discouraged, as it has been associated with bowel ischemia.
 ERCP
ERCP
 This is not indicated in mild and non-gallstone pancreatitis.
This is not indicated in mild and non-gallstone pancreatitis.
 Early (within 48 hours) ERCP is only indicated in patients with pancreatitis with concommitant cholangitis.
Early (within 48 hours) ERCP is only indicated in patients with pancreatitis with concommitant cholangitis.
 In patients without obstructive jaundice, early ERCP does not show a clear benefit and should be avoided.
In patients without obstructive jaundice, early ERCP does not show a clear benefit and should be avoided.
 Role of percutaneous or endoscopic intervention and surgery
Role of percutaneous or endoscopic intervention and surgery
 The only clear indication for intervention is infected pancreatic or peripancreatic necrosis.
The only clear indication for intervention is infected pancreatic or peripancreatic necrosis.
 Whenever infected necrosis is suspected, a CT scan with intravenous contrast is the best first test.
Whenever infected necrosis is suspected, a CT scan with intravenous contrast is the best first test.
 Infection can be proven by the presence of gas in pancreatic necrosis on CT scan or by fine needle aspiration (FNA) with Gram stain and culture of the necrosis.
Infection can be proven by the presence of gas in pancreatic necrosis on CT scan or by fine needle aspiration (FNA) with Gram stain and culture of the necrosis.
 Neither of these is 100% sensitive, and in a severely ill patient with suspected infected necrosis, intervention should be performed even in the absence of definite proof of infection.
Neither of these is 100% sensitive, and in a severely ill patient with suspected infected necrosis, intervention should be performed even in the absence of definite proof of infection.
 When infection is proven or suspected, the first intervention should be antibiotic administration (a carbapenem is standard) and percutaneous or endoscopic drainage.
When infection is proven or suspected, the first intervention should be antibiotic administration (a carbapenem is standard) and percutaneous or endoscopic drainage.
 Surgical debridement is reserved for when clinical improvement cannot be achieved by less invasive measures.
Surgical debridement is reserved for when clinical improvement cannot be achieved by less invasive measures.
 This progression from less invasive drainage procedures to more invasive surgical debridement is termed the “step-up approach.”
This progression from less invasive drainage procedures to more invasive surgical debridement is termed the “step-up approach.”
 Surgery should be delayed until at least 4 weeks after the onset of pancreatitis whenever possible.
Surgery should be delayed until at least 4 weeks after the onset of pancreatitis whenever possible.
 There are a variety of surgical techniques (open transabdominal debridement, laparoscopic debridement, video-assisted retroperitoneal debridement) with none that is clearly superior at this point.
There are a variety of surgical techniques (open transabdominal debridement, laparoscopic debridement, video-assisted retroperitoneal debridement) with none that is clearly superior at this point.
 Omission or delay of cholecystectomy in gallstone pancreatitis is associated with increased risk of recurrent attacks.
Omission or delay of cholecystectomy in gallstone pancreatitis is associated with increased risk of recurrent attacks.
 In mild to moderate pancreatitis, cholecystectomy should be performed during the index pancreatitis admission.
In mild to moderate pancreatitis, cholecystectomy should be performed during the index pancreatitis admission.
 In severe disease, cholecystectomy should be delayed at least 6 weeks, as the complication rate is high after early cholecystectomy.
In severe disease, cholecystectomy should be delayed at least 6 weeks, as the complication rate is high after early cholecystectomy.
Outcomes
 Mortality: 2% to 3% overall, may be up to 30% to 40% in SAP with organ failure
Mortality: 2% to 3% overall, may be up to 30% to 40% in SAP with organ failure
 Complications
Complications
 Systemic
Systemic
 ALI/ARDS, multiorgan failure, coagulopathy/DIC, acute renal failure (usually prerenal, confers worse progrosis)
ALI/ARDS, multiorgan failure, coagulopathy/DIC, acute renal failure (usually prerenal, confers worse progrosis)
 Severe hypocalcemia
Severe hypocalcemia
 Local/mechanical
Local/mechanical
 Acute fluid collection (30% to 50%)
Acute fluid collection (30% to 50%)
 Simple fluid accumulating in or near the pancreas within the first few days of acute pancreatitis
Simple fluid accumulating in or near the pancreas within the first few days of acute pancreatitis
 Usually resolves spontaneously and does not require treatment
Usually resolves spontaneously and does not require treatment
 Sterile pancreatic necrosis (20%)
Sterile pancreatic necrosis (20%)
 No indication for surgery if asymptomatic, no consensus for prophylactic antibiotics
No indication for surgery if asymptomatic, no consensus for prophylactic antibiotics
 Supportive measures only
Supportive measures only
 Infected pancreatic or peripancreatic necrosis (5% to 10%)
Infected pancreatic or peripancreatic necrosis (5% to 10%)
 As above, diagnosed by CT or FNA
As above, diagnosed by CT or FNA
 Usually gram-negative intestinal organisms, but may include gram positives or fungi
Usually gram-negative intestinal organisms, but may include gram positives or fungi
 Treated with antibiotics and “step up approach” as above
Treated with antibiotics and “step up approach” as above
 Carbapenems are standard antibiotic therapy.
Carbapenems are standard antibiotic therapy.
 Pseudocyst (~10%)
Pseudocyst (~10%)
 Fluid collection formed due to walled-off disruption of the pancreatic duct
Fluid collection formed due to walled-off disruption of the pancreatic duct
 Many resolve spontaneously
Many resolve spontaneously
 If large, persistent, and symptomatic should be internally drained either endoscopically or surgically
If large, persistent, and symptomatic should be internally drained either endoscopically or surgically
 Pancreatic fistulas (17% to 76% of severe cases)
Pancreatic fistulas (17% to 76% of severe cases)
 High volume and amylase content coming from the pancreatic drains are indicative of pancreatic fistula formation.
High volume and amylase content coming from the pancreatic drains are indicative of pancreatic fistula formation.
 Treatment is conservative.
Treatment is conservative.
 Endoscopic transpapillary stenting may be as safe and decrease the time to fistula closure by decompressing the pancreatitic duct.
Endoscopic transpapillary stenting may be as safe and decrease the time to fistula closure by decompressing the pancreatitic duct.
 Other
Other
 Diabetes, pleural effusion/ pancreatic ascites (think about pancreatic duct disruption), splenic/mesenteric/portal vein thrombosis with gastric varices
Diabetes, pleural effusion/ pancreatic ascites (think about pancreatic duct disruption), splenic/mesenteric/portal vein thrombosis with gastric varices
SUGGESTED READINGS
Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg. 2010;251:615-619.
Al-Omran M, Albalawi ZH, Tashkandi MF, et al. Enteral versus parenteral nutrition for acute pancreatitis. Cochrane Database Syst Rev. 2010;1:CD002837.
Bakker OJ, van Baal MC, van Santvoort HC, et al. Endoscopic transpapillary stenting or conservative treatment for pancreatic fistulas in necrotizing pancreatitis: multicenter series and literature review. Ann Surg. 2011;253:961-967.
Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101:2379-2400.
Bernhardt A, Kortgen A, Niesel H, et al. Using epidural anesthesia in patients with acute pancreatitis—prospective study of 121 patients. Anaesthesiol Reanim. 2002;27:16-22.
Besselink MG, van Santvoort HC, Buskens E, et al. Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:651-659.
Bradley EL 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg. 1993;128:586-590.
Cannon JW, Callery MP, Vollmer CM Jr. Diagnosis and management of pancreatic pseudocysts: what is the evidence? J Am Coll Surg. 2009;209:385-393.
Casaer MP, Mesotten D, Hermans G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011;365:506-517.
Eatock FC, Chong P, Menezes N, et al. A randomized study of early nasogastric versus nasojejunal feeding in severe acute pancreatitis. Am J Gastroenterol. 2005;100:432-439.
Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part III: liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134-1144.
Fagenholz PJ, Castillo CF, Harris NS, et al. Increasing United States hospital admissions for acute pancreatitis, 1988–2003. Ann Epidemiol. 2007;17:491-497.
Fagenholz PJ, Fernandez-del Castillo C, Harris NS, et al. National study of United States emergency department visits for acute pancreatitis, 1993–2003. BMC Emerg Med. 2007;7:1.
Forsmark CE, Baillie J. AGA institute technical review on acute pancreatitis. Gastroenterology. 2007;132:2022-2044.
Frey CF, Zhou H, Harvey DJ, et al. The incidence and case-fatality rates of acute biliary, alcoholic, and idiopathic pancreatitis in California, 1994–2001. Pancreas. 2006;33:336-344.
Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371: 143-152.
Gardner TB, Vege SS, Pearson RK, et al. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol. 2008;6:1070-1076.
Hernandez V, Pascual I, Almela P, et al. Recurrence of acute gallstone pancreatitis and relationship with cholecystectomy or endoscopic sphincterotomy. Am J Gastroenterol. 2004;99:2417-2423.
Jacobson BC, Vander Vliet MB, Hughes MD, et al. A prospective, randomized trial of clear liquids versus low-fat solid diet as the initial meal in mild acute pancreatitis. Clin Gastroenterol Hepatol. 2007;5:946-951; quiz 886.
Jensen EH, Borja-Cacho D, Al-Refaie WB, Vickers SM. Exocrine Pancreas. Philadelphia, PA: Elsevier Saunders; 2007.
Kutsogiannis J, Alberda C, Gramlich L, et al. Early use of supplemental parenteral nutrition in critically ill patients: results of an international multicenter observational study. Crit Care Med. 2011;39:2691-2699.
McClave SA, Dryden GW. Issues of nutritional support for the patient with acute pancreatitis. Semin Gastrointest Dis. 2002;13:154-160.
Meng WB, Li X, Li YM, et al. Three initial diets for management of mild acute pancreatitis: a meta-analysis. World J Gastroenterol. 2011;17:4235-4241.
Mofidi R, Suttie SA, Patil PV, et al. The value of procalcitonin at predicting the severity of acute pancreatitis and development of infected pancreatic necrosis: systematic review. Surgery. 2009;146:72-81.
Moraes JM, Felga GE, Chebli LA, et al. A full solid diet as the initial meal in mild acute pancreatitis is safe and result in a shorter length of hospitalization: results from a prospective, randomized, controlled, double-blind clinical trial. J Clin Gastroenterol. 2010;44:517-522.
Munsell MA, Buscaglia JM. Acute pancreatitis. J Hosp Med. 2010;5:241-250.
Petrov MS, van Santvoort HC, Besselink MG, et al. Early endoscopic retrograde cholangiopancreatography versus conservative management in acute biliary pancreatitis without cholangitis: a meta-analysis of randomized trials. Ann Surg. 2008;247:250-257.
Quan H, Wang X, Guo C. A meta-analysis of enteral nutrition and total parenteral nutrition in patients with acute pancreatitis. Gastroenterol Res Pract. 2011;2011:698248.
Sathiaraj E, Murthy S, Mansard MJ, et al. Clinical trial: oral feeding with a soft diet compared with clear liquid diet as initial meal in mild acute pancreatitis. Aliment Pharmacol Ther. 2008;28:777-781.
van Santvoort HC, Besselink MG, Bakker OJ, et al. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med. 2010;362:1491-1502.
van Santvoort HC, Besselink MG, de Vries AC, et al. Early endoscopic retrograde cholangiopancreatography in predicted severe acute biliary pancreatitis: a prospective multicenter study. Ann Surg. 2009;250: 68-75.
Vege S, Yadav D, Chari S. Pancreatitis. Malden, MA: Blackwell Publishing; 2007.
Vege SS, Gardner TB, Chari ST, et al. Low mortality and high morbidity in severe acute pancreatitis without organ failure: a case for revising the Atlanta classification to include “moderately severe acute pancreatitis.” Am J Gastroenterol. 2009;104:710-715.
Villatoro E, Mulla M, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. 2010;5:CD002941.
Wilson CT, de Moya MA. Cholecystectomy for acute gallstone pancreatitis: early vs delayed approach. Scand J Surg. 2010;99:81-85.
Wu BU, Conwell DL. Update in acute pancreatitis. Curr Gastroenterol Rep. 2010;12:83-90.
Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9:710-717, e711.
Yadav D, Lowenfels AB. Trends in the epidemiology of the first attack of acute pancreatitis: a systematic review. Pancreas. 2006;33:323-330.
4.3
Gastrointestinal Perforation
George Kasotakis
Introduction
Gastrointestinal perforation refers to complete penetration of the wall of the stomach, small or large intestine, allowing intestinal contents to flow into the peritoneal cavity.
Key Pathophysiology
 Peptic ulcer perforation occurs when the injurious effects of acid and pepsin overwhelm the mucosal barrier and acid-suppression therapy has not been instituted or is inadequate to control the local acid environment.
Peptic ulcer perforation occurs when the injurious effects of acid and pepsin overwhelm the mucosal barrier and acid-suppression therapy has not been instituted or is inadequate to control the local acid environment.
 In mechanical obstruction (obstructing tumor, volvulus, bowel herniation) without vascular compromise, ingested fluid and food, digestive secretions. and gas accumulate above the obstruction, leading to proximal bowel overdistention and eventually to microperforations or strangulating obstruction.
In mechanical obstruction (obstructing tumor, volvulus, bowel herniation) without vascular compromise, ingested fluid and food, digestive secretions. and gas accumulate above the obstruction, leading to proximal bowel overdistention and eventually to microperforations or strangulating obstruction.
 Intestinal ischemia (associated with hernia, volvulus, intussusception, severe calcific burden, or low flow states) can progress to infarction, gangrene, and perforation in as little as 6 hours.
Intestinal ischemia (associated with hernia, volvulus, intussusception, severe calcific burden, or low flow states) can progress to infarction, gangrene, and perforation in as little as 6 hours.
 Local obstruction by fecaliths (appendicitis, diverticulitis) may lead to localized luminal overdistention, compromised enteric wall blood supply, and finally severe inflammation and perforation.
Local obstruction by fecaliths (appendicitis, diverticulitis) may lead to localized luminal overdistention, compromised enteric wall blood supply, and finally severe inflammation and perforation.
 Traumatic (more commonly with penetrating injuries) or iatrogenic perforations can also occur (typically during lysis of adhesions or endoscopy).
Traumatic (more commonly with penetrating injuries) or iatrogenic perforations can also occur (typically during lysis of adhesions or endoscopy).
Common Causes to Remember
 Untreated severe peptic ulcer disease
Untreated severe peptic ulcer disease
 Malignant perforation
Malignant perforation
 Untreated appendicitis
Untreated appendicitis
 Cecal/sigmoid volvulus
Cecal/sigmoid volvulus
 Inflammatory bowel disease
Inflammatory bowel disease
 Diverticulitis
Diverticulitis
 Toxic megacolon
Toxic megacolon
 Excessive distention from bowel obstruction
Excessive distention from bowel obstruction
 Intestinal ischemia
Intestinal ischemia
 Penetrating wounds to the abdomen
Penetrating wounds to the abdomen
 Iatrogenic (most commonly during colonoscopy, ERCP, or abdominal surgery)
Iatrogenic (most commonly during colonoscopy, ERCP, or abdominal surgery)
 Symptoms and signs
Symptoms and signs
 Gastric and duodenal perforations typically present with sudden pain in the epigastrium or right upper quadrant.
Gastric and duodenal perforations typically present with sudden pain in the epigastrium or right upper quadrant.
 In contained perforated diverticulitis the symptoms are typically localized in the left lower quadrant.
In contained perforated diverticulitis the symptoms are typically localized in the left lower quadrant.
 In all other cases, location and quality of the pain depend on the part of the gastrointestinal tract that perforates.
In all other cases, location and quality of the pain depend on the part of the gastrointestinal tract that perforates.
 In general, if the perforation is contained, local pain and tenderness may be present. Systemic manifestations of acute illness are typically mild. If the leakage of bowel contents is not contained, diffuse abdominal pain worsened by movement and associated with rigidity, guarding, and rebound tenderness is typically present. Fever and tachycardia are common.
In general, if the perforation is contained, local pain and tenderness may be present. Systemic manifestations of acute illness are typically mild. If the leakage of bowel contents is not contained, diffuse abdominal pain worsened by movement and associated with rigidity, guarding, and rebound tenderness is typically present. Fever and tachycardia are common.
Diagnosis
 Peritoneal signs (diffuse abdominal pain, rigidity, guarding, and rebound tenderness) should alert the clinical staff for the possibility of bowel perforation. Fever, leukocytosis, and tachycardia are frequently present.
Peritoneal signs (diffuse abdominal pain, rigidity, guarding, and rebound tenderness) should alert the clinical staff for the possibility of bowel perforation. Fever, leukocytosis, and tachycardia are frequently present.
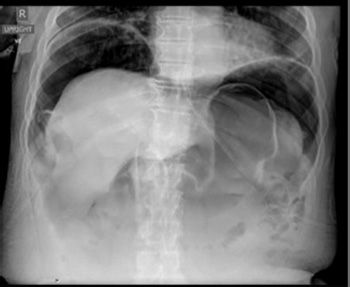
Figure 4.3.1 Free air under the diaphragm after iatrogenic duodenal perforation.
 Free air in the subdiaphragmatic space can be identified on plain x-rays (Fig. 4.3.1).
Free air in the subdiaphragmatic space can be identified on plain x-rays (Fig. 4.3.1).
 Abdominal CT may provide information about the location of the perforation (localized bowel wall thickening, mesenteric and soft tissue stranding adjacent to the perforation) (Fig. 4.3.2).
Abdominal CT may provide information about the location of the perforation (localized bowel wall thickening, mesenteric and soft tissue stranding adjacent to the perforation) (Fig. 4.3.2).
Management and Treatment
 Typically depends on the host’s physiology and whether the perforation is contained or not.
Typically depends on the host’s physiology and whether the perforation is contained or not.
 Broad-spectrum antibiotics, bowel rest, and percutaneous drainage of extraluminal collections may be used for the clinically well patient with localized peritoneal irritation.
Broad-spectrum antibiotics, bowel rest, and percutaneous drainage of extraluminal collections may be used for the clinically well patient with localized peritoneal irritation.
 In disseminated peritonitis, surgical exploration with cavity washout is usually warranted. Perforations may be patched (gastroduodenal), primarily repaired (intestinal), excised (gastric, intestinal), or proximally diverted (diverticulitis).
In disseminated peritonitis, surgical exploration with cavity washout is usually warranted. Perforations may be patched (gastroduodenal), primarily repaired (intestinal), excised (gastric, intestinal), or proximally diverted (diverticulitis).
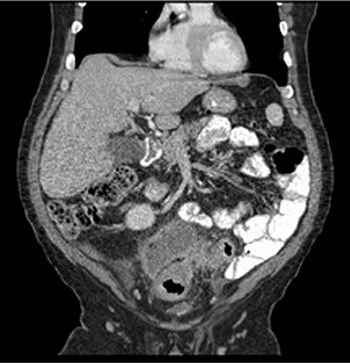
Figure 4.3.2 Localized sigmoid wall thickening, adjacent soft tissue stranding, and abscess in the lower abdomen with acute diverticulitis.
SUGGESTED READINGS
Beckham H, Whitlow CB. The medical and nonoperative treatment of diverticulitis. Clin Colon Rectal Surg. 2009;22(3):155-160.
Castellví J, Pi F, Sueiras A, et al. Colonoscopic perforation: useful parameters for early diagnosis and conservative treatment. Int J Colorectal Dis. 2011;26(9):1183-1190.
Horn AE, Ufberg JW. Appendicitis, diverticulitis and colitis. Emerg Med Clin North Am. 2011;29(2):347-368.
Rafferty J, Shellito P, Hyman NH, et al. Practice parameters for sigmoid diverticulitis. Dis Colon Rectum. 2006;49(7):939-944.
Spirt MJ. Complicated intra-abdominal infections: a focus on appendicitis and diverticulitis. Postgrad Med. 2012;122(1):39.
4.4
Acute Mesenteric Ischemia
Haytham M. A. Kaafarani
Introduction
 Acute mesenteric ischemia (AMI) is caused by a sudden decrease in perfusion to the small or large bowel, resulting in a mismatch of metabolic supply and demand, with an ultimate risk of bowel necrosis.
Acute mesenteric ischemia (AMI) is caused by a sudden decrease in perfusion to the small or large bowel, resulting in a mismatch of metabolic supply and demand, with an ultimate risk of bowel necrosis.
Epidemiology
 AMI is rare: its incidence increases with age and is reported to be around 1.09 per 100,000 person-years.
AMI is rare: its incidence increases with age and is reported to be around 1.09 per 100,000 person-years.
 AMI is most common after the sixth decade of life, especially in patients with peripheral or coronary atherosclerosis.
AMI is most common after the sixth decade of life, especially in patients with peripheral or coronary atherosclerosis.
Key Pathophysiology
 The mesenteric blood supply is derived from three major arteries:
The mesenteric blood supply is derived from three major arteries:
 The celiac artery
The celiac artery
 The superior mesenteric artery (SMA = most commonly implicated vessel)
The superior mesenteric artery (SMA = most commonly implicated vessel)
 The inferior mesenteric artery (IMA)
The inferior mesenteric artery (IMA)
 The mesenteric arterial anatomy includes extensive collateral circulation connecting the major mesenteric vessels to each other as well as to the systemic circulation.
The mesenteric arterial anatomy includes extensive collateral circulation connecting the major mesenteric vessels to each other as well as to the systemic circulation.
 The probability of intestinal ischemia depends upon the adequacy of systemic and collateral perfusion, the number and caliber of affected vessels, and the duration of the insult.
The probability of intestinal ischemia depends upon the adequacy of systemic and collateral perfusion, the number and caliber of affected vessels, and the duration of the insult.
 The mesenteric vasculature can compensate for up to 75% acute reduction in perfusion for as long as 12 hours through increased oxygen extraction at the tissue level and increased collateral circulation.
The mesenteric vasculature can compensate for up to 75% acute reduction in perfusion for as long as 12 hours through increased oxygen extraction at the tissue level and increased collateral circulation.
 Despite the extensive collateral mesenteric circulation, prolonged bowel hypoperfusion results in vasoconstriction, thus ultimately reducing collateral flow.
Despite the extensive collateral mesenteric circulation, prolonged bowel hypoperfusion results in vasoconstriction, thus ultimately reducing collateral flow.
 Ischemic damage is due to both hypoxic and reperfusion injury, often with vasoconstriction persisting after restoration of blood flow.
Ischemic damage is due to both hypoxic and reperfusion injury, often with vasoconstriction persisting after restoration of blood flow.
 Ischemic injury to the bowel results in the systemic release of intracellular breakdown products and an increase in anaerobic metabolism.
Ischemic injury to the bowel results in the systemic release of intracellular breakdown products and an increase in anaerobic metabolism.
Etiology and Risk Factors
 The etiology of AMI may be categorized as
The etiology of AMI may be categorized as
 Mesenteric arterial embolism
Mesenteric arterial embolism
 Mesenteric arterial thrombosis
Mesenteric arterial thrombosis
 Nonocclusive mesenteric ischemia
Nonocclusive mesenteric ischemia
 Mesenteric venous thrombosis
Mesenteric venous thrombosis
 Mesenteric arterial embolism
Mesenteric arterial embolism
 40% to 50% of cases
40% to 50% of cases
 Most common cause: embolization from cardiac source to SMA
Most common cause: embolization from cardiac source to SMA
 Occlusion usually at a branch of the SMA just distal to the middle colic artery, resulting in relative sparing of the proximal jejunum and distal large bowel
Occlusion usually at a branch of the SMA just distal to the middle colic artery, resulting in relative sparing of the proximal jejunum and distal large bowel
 Risk factors
Risk factors
 Cardiac arrhythmia (e.g., atrial fibrillation)
Cardiac arrhythmia (e.g., atrial fibrillation)
 Myocardial infarction/ventricular aneurysm
Myocardial infarction/ventricular aneurysm
 Recent cardiac or aortic catheterization
Recent cardiac or aortic catheterization
 Recent cardiac valve replacement
Recent cardiac valve replacement
 Endocarditis
Endocarditis
 Atrial septal defect (resulting in paradoxical embolus)
Atrial septal defect (resulting in paradoxical embolus)
 Mesenteric arterial thrombosis
Mesenteric arterial thrombosis
 25% of cases
25% of cases
 Most common cause is atherosclerosis
Most common cause is atherosclerosis
 Occlusion usually at the origin of the mesenteric vessel (SMA or celiac)
Occlusion usually at the origin of the mesenteric vessel (SMA or celiac)
 Less common causes include arterial dissection, arterial aneurysm, vasculitis, and iatrogenic injury (i.e., intraoperative)
Less common causes include arterial dissection, arterial aneurysm, vasculitis, and iatrogenic injury (i.e., intraoperative)
 Risk factors
Risk factors
 Known atherosclerosis/vascular disease
Known atherosclerosis/vascular disease
 Abdominal trauma
Abdominal trauma
 Infection
Infection
 Nonocclusive mesenteric ischemia
Nonocclusive mesenteric ischemia
 20% of cases
20% of cases
 Occurs most commonly due to a global reduction in cardiac output
Occurs most commonly due to a global reduction in cardiac output
 Risk factors
Risk factors
 Hypovolemia
Hypovolemia
 Sepsis
Sepsis
 Aortic insufficiency
Aortic insufficiency
 Status post cardiac surgery
Status post cardiac surgery
 Digoxin use (may result in splanchnic vasoconstriction)
Digoxin use (may result in splanchnic vasoconstriction)
 Cocaine abuse
Cocaine abuse
 Mesenteric venous thrombosis
Mesenteric venous thrombosis
 5% to 10% of cases
5% to 10% of cases
 Usually associated with the existence of a hypercoagulable state
Usually associated with the existence of a hypercoagulable state
 Risk factors
Risk factors
 Malignancy
Malignancy
 Inherited coagulopathy (i.e., Factor V Leiden, Factor C, or Factor S deficiency)
Inherited coagulopathy (i.e., Factor V Leiden, Factor C, or Factor S deficiency)
 Oral contraceptive use
Oral contraceptive use
 Portal hypertension
Portal hypertension
 Nephrotic syndrome
Nephrotic syndrome
 History of prior deep venous thrombosis or pulmonary embolus
History of prior deep venous thrombosis or pulmonary embolus
Diagnosis
 History and physical exam
History and physical exam
 History and physical exam alone are notoriously nonsensitive and nonspecific for AMI.
History and physical exam alone are notoriously nonsensitive and nonspecific for AMI.
 Embolic AMI: sudden onset of severe periumbilical pain, typically out of proportion to physical exam findings
Embolic AMI: sudden onset of severe periumbilical pain, typically out of proportion to physical exam findings
 Arterial thrombosis or nonocclusive ischemia, however, can occur more insidiously with 95% of patients reporting a 24-hour history of vague abdominal pain.
Arterial thrombosis or nonocclusive ischemia, however, can occur more insidiously with 95% of patients reporting a 24-hour history of vague abdominal pain.
 May also be accompanied by anorexia, vomiting, and diarrhea
May also be accompanied by anorexia, vomiting, and diarrhea
 Patients with transmural bowel necrosis can present with peritonitis, sepsis, and shock.
Patients with transmural bowel necrosis can present with peritonitis, sepsis, and shock.
 Laboratories
Laboratories
 No single serologic value is diagnostic or prognostic, but a common presentation includes:
No single serologic value is diagnostic or prognostic, but a common presentation includes:
 Leukocytosis with left shift
Leukocytosis with left shift
 Hemoconcentration
Hemoconcentration
 Anion gap metabolic acidosis (lactic acidosis)
Anion gap metabolic acidosis (lactic acidosis)
 Hyperamylasemia
Hyperamylasemia
 Elevated D-lactate
Elevated D-lactate
 Usefulness is controversial
Usefulness is controversial
 Estimated sensitivity of 70% to 100%
Estimated sensitivity of 70% to 100%
 Estimated specificity of 40% to 90%
Estimated specificity of 40% to 90%
 Improves with exclusion of shock, diabetic ketoacidosis, renal failure, and hepatic failure
Improves with exclusion of shock, diabetic ketoacidosis, renal failure, and hepatic failure
 Imaging studies
Imaging studies
 Plain abdominal radiographs
Plain abdominal radiographs
 Nonspecific; normal in up to 25% of patients with ischemic bowel
Nonspecific; normal in up to 25% of patients with ischemic bowel
 Significant findings include “thumbprinting” suggestive of intestinal wall edema, pneumatosis intestinalis suggestive of bowel wall necrosis, or pneumoperitoneum suggestive of bowel perforation
Significant findings include “thumbprinting” suggestive of intestinal wall edema, pneumatosis intestinalis suggestive of bowel wall necrosis, or pneumoperitoneum suggestive of bowel perforation
 Doppler-flow ultrasonography
Doppler-flow ultrasonography
 Used in detection of stenosis or occlusion of mesenteric vessels (does not detect low-flow etiologies)
Used in detection of stenosis or occlusion of mesenteric vessels (does not detect low-flow etiologies)
 Often technically limited due to interference from air-filled bowel
Often technically limited due to interference from air-filled bowel
 Abdominal computed tomographic angiography (CTA)
Abdominal computed tomographic angiography (CTA)
 Allows visualization of bowel wall thickening, pneumatosis intestinalis, portal venous gas and arterial occlusions, with both sensitivity and specificity above 90%
Allows visualization of bowel wall thickening, pneumatosis intestinalis, portal venous gas and arterial occlusions, with both sensitivity and specificity above 90%
 Higher accuracy for thromboembolic etiologies versus nonocclusive ischemia
Higher accuracy for thromboembolic etiologies versus nonocclusive ischemia
 Can also be used to rule out other causes of acute abdomen
Can also be used to rule out other causes of acute abdomen
 Mesenteric angiography
Mesenteric angiography
 Early angiography (within 12 hours of event) associated with improved survival
Early angiography (within 12 hours of event) associated with improved survival
 Allows simultaneous intervention to restore blood flow
Allows simultaneous intervention to restore blood flow
 Magnetic resonance angiography (MRA)
Magnetic resonance angiography (MRA)
 More detailed information about mesenteric vessels
More detailed information about mesenteric vessels
 More research needed to compare this modality to conventional and CT angiography.
More research needed to compare this modality to conventional and CT angiography.
 Intraoperative diagnosis
Intraoperative diagnosis
 Exploratory laparotomy is the gold standard and .zrequired for suspicion of any bowel infarction.
Exploratory laparotomy is the gold standard and .zrequired for suspicion of any bowel infarction.
Treatment and Management
 Goal of treatment is restoration of mesenteric perfusion.
Goal of treatment is restoration of mesenteric perfusion.
 Aggressive hemodynamic monitoring and support (isotonic fluids and/or vasopressors)
Aggressive hemodynamic monitoring and support (isotonic fluids and/or vasopressors)
 Broad-spectrum IV antibiotics
Broad-spectrum IV antibiotics
 Immediate therapeutic intravenous anticoagulation
Immediate therapeutic intravenous anticoagulation
 Correction of electrolyte and acid–base abnormalities
Correction of electrolyte and acid–base abnormalities
 Mesenteric arteriography
Mesenteric arteriography
 Hypotension or hypovolemia may lead to false positive studies.
Hypotension or hypovolemia may lead to false positive studies.
 Therapeutic options include intraarterial vasodilators (papaverine), thrombolytic agents, angioplasty, stenting, and embolectomy.
Therapeutic options include intraarterial vasodilators (papaverine), thrombolytic agents, angioplasty, stenting, and embolectomy.
 Consider surgery for emergent situations when there is suspicion of intestinal infarction or perforation.
Consider surgery for emergent situations when there is suspicion of intestinal infarction or perforation.
 Mesenteric arterial embolism
Mesenteric arterial embolism
 Surgical embolectomy versus local thrombolytic therapy
Surgical embolectomy versus local thrombolytic therapy
 Surgical embolectomy
Surgical embolectomy
 Arteriotomy distal to embolus followed by advancement of balloon-tipped embolectomy (Fogarty) catheter
Arteriotomy distal to embolus followed by advancement of balloon-tipped embolectomy (Fogarty) catheter
 Examination for infarcted bowel 20 to 30 minutes following revascularization
Examination for infarcted bowel 20 to 30 minutes following revascularization
 Second look laparotomy within 24 to 48 hours
Second look laparotomy within 24 to 48 hours
 Local thrombolytic therapy
Local thrombolytic therapy
 May be appropriate shortly after onset of symptoms if there is no clinical evidence of bowel infarction, and no contraindication to thrombolysis (tissue plasminogen activator, streptokinase, etc.)
May be appropriate shortly after onset of symptoms if there is no clinical evidence of bowel infarction, and no contraindication to thrombolysis (tissue plasminogen activator, streptokinase, etc.)
 >90% angiographic resolution of SMA occlusion with thrombolysis
>90% angiographic resolution of SMA occlusion with thrombolysis
 Surgical intervention mandatory if no resolution within several hours or with worsening clinical status
Surgical intervention mandatory if no resolution within several hours or with worsening clinical status
 Long-term anticoagulation to prevent future embolic events
Long-term anticoagulation to prevent future embolic events
 Mesenteric arterial thrombosis
Mesenteric arterial thrombosis
 Treatment is primarily surgical.
Treatment is primarily surgical.
 Nonsurgical management with heparin anticoagulation is only in the absence of bowel infarction and with angiographic evidence of sufficient collateral perfusion.
Nonsurgical management with heparin anticoagulation is only in the absence of bowel infarction and with angiographic evidence of sufficient collateral perfusion.
 Thrombectomy alone is unlikely to result in durable response given widespread atherosclerotic lesions. Therefore, combination of thrombectomy, revascularization techniques, and nonviable bowel resection is typically needed.
Thrombectomy alone is unlikely to result in durable response given widespread atherosclerotic lesions. Therefore, combination of thrombectomy, revascularization techniques, and nonviable bowel resection is typically needed.
 Arterial revascularization (open surgical visceral bypass) is associated with a high perioperative mortality (approximately 50%) but good long-term patency.
Arterial revascularization (open surgical visceral bypass) is associated with a high perioperative mortality (approximately 50%) but good long-term patency.
 Endovascular stenting has been also reported in small case series.
Endovascular stenting has been also reported in small case series.
 Long-term antiplatelet agents may reduce risk of recurrence.
Long-term antiplatelet agents may reduce risk of recurrence.
 Mesenteric venous thrombosis
Mesenteric venous thrombosis
 Initial management with heparin anticoagulation and workup for a hypercoagulable state
Initial management with heparin anticoagulation and workup for a hypercoagulable state
 Close observation with serial abdominal exams is warranted; prompt laparotomy is necessary for any suspicion of infarcted bowel.
Close observation with serial abdominal exams is warranted; prompt laparotomy is necessary for any suspicion of infarcted bowel.
 Small reports of successful venous thrombolysis; insufficient data to recommend thrombolysis over standard therapy
Small reports of successful venous thrombolysis; insufficient data to recommend thrombolysis over standard therapy
 Prevention of recurrence with long-term anticoagulation
Prevention of recurrence with long-term anticoagulation
 Nonocclusive mesenteric ischemia
Nonocclusive mesenteric ischemia
 Primary therapy is reversal of the underlying condition resulting in the low cardiac output state.
Primary therapy is reversal of the underlying condition resulting in the low cardiac output state.
 Successful use of local intraarterial angiographic papaverine infusion has been reported: may also be indicated postoperatively for 24 to 48 hours.
Successful use of local intraarterial angiographic papaverine infusion has been reported: may also be indicated postoperatively for 24 to 48 hours.
 Long-term management with standard-dose aspirin.
Long-term management with standard-dose aspirin.
Outcomes
 Reported mortality approximates 60% to 70%.
Reported mortality approximates 60% to 70%.
 Range is from 20% with early diagnosis and management to greater than 70% following bowel infarction.
Range is from 20% with early diagnosis and management to greater than 70% following bowel infarction.
 Diagnosis prior to infarction is the single strongest predictor of survival.
Diagnosis prior to infarction is the single strongest predictor of survival.
 Other predictors of mortality are advanced age, poor functional status, and prior cardiac surgery.
Other predictors of mortality are advanced age, poor functional status, and prior cardiac surgery.
 Reported 30-day postoperative morbidity approximates 60%.
Reported 30-day postoperative morbidity approximates 60%.
 Pulmonary and infectious complications are the most common.
Pulmonary and infectious complications are the most common.
SUGGESTED READINGS
Boley SJ, Brandt LJ, Sammartano RJ. History of mesenteric ischemia. The evolution of a diagnosis and management. Surg Clin North Am. 1997;77(2):275.
Cho JS, Carr JA, Jacobsen G, et al. Long-term outcome after mesenteric artery reconstruction: a 37-year experience. J Vasc Surg. 2002;35(3): 453.
Demirpolat G, Oran I, Tamsel S, et al. Acute mesenteric ischemia: endovascular therapy. Abdom Imaging. 2007;32(3):299.
Evennett NJ, Petrov MS, Mittal A, et al. Systematic review and pooled estimates for the diagnostic accuracy of serological markers for intestinal ischemia. World J Surg. 2009;33(7):1374-1383.
Gupta PK, Natarajan B, Gupta H, et al. Morbidity and mortality after bowel resection for acute mesenteric ischemia. Surgery. 2011;150(4):779-787.
Huerta C, Rivero E, Montoro MA, et al. Risk factors for intestinal ischaemia among patients registered in a UK primary care database: a nested case-control study. Aliment Pharmacol Ther. 2011;33(8):969-978.
Klein HM, Lensing R, Klosterhalfen B, et al. Diagnostic imaging of mesenteric infarction. Radiology. 1995;197:79.
Kumar, S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med. 2001;345(23):1683.
McKinsey JF, Gewertz BL. Acute mesenteric ischemia. Surg Clin North Am. 1997;77:307.
Ofer A, Abadi S, Nitecki S, et al. Multidetector CT angiography in the evaluation of acute mesenteric ischemia. Eur Radiol. 2009;19(1):24-30.
Oldenburg WA, Lau LL, Rodenberg TJ, et al. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164:1054-1062.
Park WM, Gloviczki P, Cherry KJ Jr, et al. Contemporary management of acute mesenteric ischemia: factors associated with survival. J Vasc Surg. 2002;35(3):445-452.
Schoots IG, Levi MM, Reekers JA, et al. Thrombolytic therapy for acute superior mesenteric artery occlusion. J Vasc Interv Radiol. 2005;16(3):317.
Vicente DC, Kazmers A. Acute mesenteric ischemia. Curr Opin Cardiol. 1999;14(5):453-458.
Wyers MC, Powell RJ, Nolan BW, et al. Retrograde mesenteric stenting during laparotomy for acute occlusive mesenteric ischemia. J Vasc Surg. 2007;45(2):269.
4.5
Gastrointestinal Bleeding
Kathryn L. Butler
Introduction
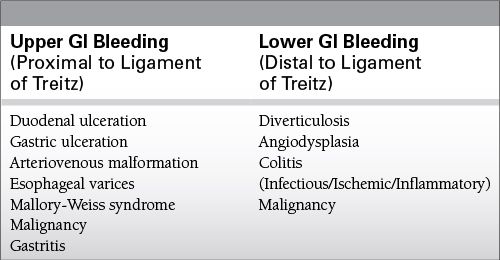
 Upper gastrointestinal (GI) bleeding accounts for 80% of cases.
Upper gastrointestinal (GI) bleeding accounts for 80% of cases.
 Jejunal/ileal bleeding is responsible for <5% of cases.
Jejunal/ileal bleeding is responsible for <5% of cases.
 Remember vascular sources in postoperative patients
Remember vascular sources in postoperative patients
 Visceral artery pseudoaneurysm after foregut surgery
Visceral artery pseudoaneurysm after foregut surgery
 Aortoenteric fistula after aortic aneurysm repair
Aortoenteric fistula after aortic aneurysm repair
Common Causes to Remember
Epidemiology
 Annual incidence 170 cases per 100,000 adults
Annual incidence 170 cases per 100,000 adults
 Incidence increases with advancing age
Incidence increases with advancing age
 More common in men than women
More common in men than women
Key Pathophysiology
 Upper GI bleeding
Upper GI bleeding
 Responsible for 80% of acute bleeds
Responsible for 80% of acute bleeds
 Melena
Melena
 Black, tarry schools
Black, tarry schools
 Usually from gastric acid degradation of hemoglobin to hematin
Usually from gastric acid degradation of hemoglobin to hematin
 Can also occur secondary to digestive enzyme action in small intestine
Can also occur secondary to digestive enzyme action in small intestine
 Hematemesis
Hematemesis
 Can be bright red or coffee grounds
Can be bright red or coffee grounds
 Brisk upper GI bleeding can present as hematochezia (bright red blood per rectum) without hematemesis or melena.
Brisk upper GI bleeding can present as hematochezia (bright red blood per rectum) without hematemesis or melena.
 Most common causes: peptic ulcer disease (PUD) and varices
Most common causes: peptic ulcer disease (PUD) and varices
 PUD
PUD
 30% to 50% of upper GI bleeding cases
30% to 50% of upper GI bleeding cases
 Bleeding most frequent indication for operation, and primary cause of death
Bleeding most frequent indication for operation, and primary cause of death
 Significant bleeding from erosion into artery of submucosa or larger vessel
Significant bleeding from erosion into artery of submucosa or larger vessel
 Highest morbidity from erosion into gastroduodenal or left gastric arteries
Highest morbidity from erosion into gastroduodenal or left gastric arteries
 Duodenal ulcers more common than gastric
Duodenal ulcers more common than gastric
 Causes include Helicobacter pylori and nonsteroidal anti-inflammatory drugs (NSAIDs)
Causes include Helicobacter pylori and nonsteroidal anti-inflammatory drugs (NSAIDs)
 Varices
Varices
 20% of upper GI bleeding cases
20% of upper GI bleeding cases
 Secondary to portal hypertension
Secondary to portal hypertension
 Compared with nonvariceal bleeding, there is increased mortality, risk of rebleeding, and need for transfusions.
Compared with nonvariceal bleeding, there is increased mortality, risk of rebleeding, and need for transfusions.
 Gastroesophageal (GE) varices most common
Gastroesophageal (GE) varices most common
 Although nonvariceal sources still account for most bleeds in cirrhotic patients, given high mortality rate of variceal bleeding, treat empirically for variceal hemorrhage (octreotide) until endoscopy identifies another source.
Although nonvariceal sources still account for most bleeds in cirrhotic patients, given high mortality rate of variceal bleeding, treat empirically for variceal hemorrhage (octreotide) until endoscopy identifies another source.
 Mallory-Weiss syndrome
Mallory-Weiss syndrome
 5% to 10% of upper GI bleeding cases
5% to 10% of upper GI bleeding cases
 Mucosal and submucosal tears near GE junction, usually along lesser curvature
Mucosal and submucosal tears near GE junction, usually along lesser curvature
 Typically in alcoholic patients, following period of intense retching and vomiting
Typically in alcoholic patients, following period of intense retching and vomiting
 Supportive therapy usually adequate
Supportive therapy usually adequate
 90% of cases self-limited
90% of cases self-limited
 Mucosa heals within 72 hours.
Mucosa heals within 72 hours.
 Gastritis
Gastritis
 Multiple superficial erosions in gastric mucosa
Multiple superficial erosions in gastric mucosa
 Occurs secondary to acid injury combined with ischemia from hypoperfused states (“stress gastritis”)
Occurs secondary to acid injury combined with ischemia from hypoperfused states (“stress gastritis”)
 Low risk of significant bleeding
Low risk of significant bleeding
 Risk factors
Risk factors
 Ventilator dependence > 48 hours
Ventilator dependence > 48 hours
 Coagulopathy
Coagulopathy
 Head injury
Head injury
 Prophylactic treatment in at-risk patients: H2 blockers, proton pump inhibitors, or sucralfate
Prophylactic treatment in at-risk patients: H2 blockers, proton pump inhibitors, or sucralfate
 Lower GI bleeding
Lower GI bleeding
 Colonic source in 95% of patients
Colonic source in 95% of patients
 Presents with hematochezia
Presents with hematochezia
 Melena if very slow bleed or from proximal source
Melena if very slow bleed or from proximal source
 Incidence increases with age
Incidence increases with age
 Typically less severe, more intermittent bleeding than upper GI bleeding
Typically less severe, more intermittent bleeding than upper GI bleeding
 In 40% of patients, more than one potential source identified
In 40% of patients, more than one potential source identified
 Diverticulosis
Diverticulosis
 30% to 40% of cases
30% to 40% of cases
 Bleeding through vasa recti as they penetrate submucosa
Bleeding through vasa recti as they penetrate submucosa
 75% of bleeds stop spontaneously
75% of bleeds stop spontaneously
 10% rebleed in a year, 50% rebleed in 10 years
10% rebleed in a year, 50% rebleed in 10 years
 More common in left colon, but right-sided disease responsible for 50% of bleeding
More common in left colon, but right-sided disease responsible for 50% of bleeding
 Angiodysplasia
Angiodysplasia
 Acquired, degenerative lesions secondary to progressive dilation of normal blood vessels in the submucosa
Acquired, degenerative lesions secondary to progressive dilation of normal blood vessels in the submucosa
 Most common in patients > 50 years
Most common in patients > 50 years
 Neoplasia
Neoplasia
 Usually slow, chronic bleeding
Usually slow, chronic bleeding
 Must consider cancer in all cases of lower GI bleed
Must consider cancer in all cases of lower GI bleed
 Anorectal disease
Anorectal disease
 Anal fissure
Anal fissure
 Internal hemorrhoids
Internal hemorrhoids
 5% to 10% of lower GI bleeds
5% to 10% of lower GI bleeds
 Imperative to rule out other sources before attributing a lower GI bleed to anorectal disease
Imperative to rule out other sources before attributing a lower GI bleed to anorectal disease
 Colitis/Enteritis
Colitis/Enteritis
 Ischemic
Ischemic
 Occlusive
Occlusive
 Arterial thrombus/embolus
Arterial thrombus/embolus
 Venous thrombus
Venous thrombus
 Nonocclusive
Nonocclusive
 Low-flow states
Low-flow states
 Infectious
Infectious
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


