Edited by Anthony Brown Michael J Gingold, Adam B Bystrzycki and Flavia M Cicuttini Rheumatological conditions are common and encompass inflammatory or connective tissue diseases and mechanical/musculoskeletal conditions. Life-threatening emergencies are rare and relate to either the underlying condition or its treatment. The challenge is in making the distinction between the two, as treatment is frequently diametrically opposed; for example, the difference between administering further immunosuppression and giving antibiotics. The most common rheumatological emergency seen in the emergency department (ED) is acute monoarthritis (see Chapter 14.2). This chapter discusses the important general emergencies associated with rheumatological conditions. Many of these are multisystem diseases and emergencies relate to either a primary joint problem, an extra-articular manifestation or sometimes to the drugs used in management. Many of these conditions are autoimmune, thus immunosuppression is usually central to their management making infection a frequent complication. More targeted, so-called biological therapies, which inhibit proinflammatory cytokines as well as B- and T-cell activity have been developed. They carry their own set of potential complications, again including infection. Rheumatoid arthritis (RA) affects 1–2% of the population across most ethnic subgroups and is two to three times more common in females than males. RA is a systemic inflammatory condition of unknown aetiology characterized by widespread synovitis resulting in joint erosions and destruction. It may also produce extra-articular manifestations including vasculitis and visceral involvement. Management typically involves symptom relief with non-steroidal anti-inflammatory drugs (NSAIDs) and/or corticosteroids and early initiation of conventional disease modifying antirheumatic drugs (DMARDs). These include methotrexate, leflunomide, sulphasalazine and hydroxychloroquine and, if these agents fail to control disease progression, biological agents are commenced. The latter act by inhibiting tumour necrosis factor-α (TNF-α) (infliximab, etanercept, adalimumab, golimumab, certolizumab pegol), interleukin-6 (IL-6) (tocilizumab), T-cell co-stimulation (abatacept) or by depleting B cells (rituximab). See Chapter 14.3 for further details on the clinical features of RA, its diagnosis, investigation and management. A patient with established RA may present with an acutely painful, hot, swollen joint that may be due to the underlying condition or, alternatively it may be due to a septic arthritis. Patients with RA are two to three times more susceptible than matched controls [1]. The risk is approximately twofold higher again in RA patients on TNF-α inhibitors compared to RA patients on conventional DMARDs [2]. Thus, the possibility of septic arthritis must be considered in a patient with RA who has acute monoarthritis out of keeping with their disease activity. See Chapter 14.2 for an approach to acute monoarthritis. Cervical spine involvement in RA is common with a prevalence of up to 61%. It is more likely in those with long-standing, erosive disease and disease of greater severity and activity [3]. Cervical spine involvement is associated with increased mortality [4] and may manifest as atlanto-axial subluxation (most commonly anterior movement on the axis) or subluxation of lower cervical vertebrae. Either of these can result in cervical myelopathy. Cervical spine subluxation is often asymptomatic – up to 44% in one study [3]. The most common symptom is neck pain that may radiate towards the occiput. Other suggestive symptoms include sensory loss in hands or feet, paraesthesia or weakness in the distribution of cervical nerve roots and slowly progressive spastic quadriparesis. Important ‘red flags’ suggesting cervical myelopathy are listed in Table 14.1.1. Table 14.1.1 Symptoms and signs of cervical myelopathy Symptoms Pain Weakness Peripheral paraesthesia Gait disturbance Sphincter dysfunction Changes in consciousness Respiratory dysfunction Signs Spasticity Weakness Hyperreflexia of deep tendon reflexes Extensor plantar response Gait ataxia Respiratory irregularity Plain X-rays of the cervical spine (lateral view) may demonstrate an increase in separation between the odontoid and arch of C1. Prior to taking flexion–extension films, perform plain ‘peg’ X-rays through the open mouth to exclude odontoid fracture or severe atlanto-axial subluxation. Computed tomography (CT) can provide additional useful information, although if there is concern regarding myelopathy, magnetic resonance imaging (MRI) is more sensitive. The main implication of RA of the cervical spine in the ED is when endotracheal intubation is required. Excessive manipulation (neck flexion with head extension) of the rheumatoid cervical spine in preparation for intubation can result in significant morbidity and even mortality from atlanto-axial and subaxial subluxation. Whenever possible, a patient with RA who is scheduled to undergo surgery should have imaging of his or her cervical spine prior to endotracheal intubation. Anaesthetic consultation is recommended. Patients with subluxation and signs of spinal cord compression represent a neurosurgical emergency and prompt referral is essential. Vasculitis in RA can occur in both small- and medium-sized vessels. Patients typically have long-standing, aggressive joint disease. This presentation, although important, is becoming less frequent. Rheumatoid vasculitis presentations are varied and non-specific. Patients frequently have constitutional symptoms and fatigue. The most common manifestation is cutaneous vasculitis with deep skin ulcers on the lower limbs [5], digital ischaemia and gangrene (medium vessels) or palpable purpura (small vessels). Mononeuritis multiplex is another frequent presentation resulting from vasculitic infarction of the vasa nervorum, which typically has an acute onset. Medium vessel rheumatoid vasculitis may cause organ infarction and necrosis. Rheumatoid vasculitis can mimic polyarteritis nodosa (PAN) with involvement of the renal arteries and, less commonly, the mesenteric circulation. Pericarditis may accompany rheumatoid vasculitis, but coronary vasculitis is rare. Ocular manifestations include episcleritis and peripheral ulcerative keratitis. Central nervous system (CNS) involvement is rare. Rheumatoid factor titre is typically elevated in rheumatoid vasculitis, although this is a non-specific finding. Rheumatoid vasculitis in the absence of rheumatoid factor is rare. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are also elevated. Check a full blood count, urea and electrolytes and a midstream urine specimen for active urinary sediment including abnormal red cells or casts, as well as infection. Further investigations are directed at the relevant organ system involved usually after specialist consultation: Angiography findings are non-specific and not always diagnostic. Suspect rheumatoid vasculitis in patients with a long-standing history of seropositive RA who presents with constitutional symptoms and one of the above features, such as a typical rash, digital gangrene, red eye, neurological complaint or an active urinary sediment. Systemic rheumatoid vasculitis has a poor prognosis without immune-suppressive therapy. Urgent rheumatology consultation is required as treatment usually consists of high-dose corticosteroids as well as cyclophosphamide, often necessitating hospital admission. Pulmonary manifestations include pleural-based disease, such as pleurisy or pleural effusions, or parenchymal disease, such as interstitial lung disease (the most common manifestation), organizing pneumonia and rheumatoid nodules. Caplan’s syndrome occurs when RA is associated with pneumoconiosis. Important differential diagnoses include infection due to immune suppression, treatment-related toxicity, such as methotrexate-induced pneumonitis, and other medical co-morbidities including chronic obstructive pulmonary disease. Pleural disease often resolves without treatment, although an NSAID may be used symptomatically. Parenchymal disease documented on chest X-ray or high-resolution CT requires specialist treatment. Pericarditis occurs in 30% of RA patients based on echocardiography, but less than 10% have clinical features. It generally presents when there is active joint and other extra-articular disease and management consists of NSAIDs or prednisolone. Myocarditis is a rare manifestation of RA. It may be granulomatous and, depending on its location, can produce valvular (especially mitral) incompetence or conduction defects. Sjögren’s syndrome may present in a primary form as a systemic disease, but can also occur secondary to RA and other connective tissue disorders. The classic symptoms are dry gritty eyes, dry mouth or both. Treatment is usually symptomatic in patients with no other features. Felty’s syndrome is characterized by seropositive RA, splenomegaly and neutropaenia. There may be other cytopaenias, as well as leg ulcers, and infection is a risk. Renal involvement with RA is rare and includes vasculitis and glomerulonephritis. Secondary amyloidosis can occur in patients with long-standing active disease. However, many medications used in RA are nephrotoxic, in particular NSAIDs and ciclosporin. Vasculitis may produce mononeuritis multiplex, otherwise central nervous system involvement is rare. Patients with RA and other connective tissue diseases, such as systemic lupus erythematosus (SLE), have an increased risk of ischaemic heart disease (IHD) [6]. This occurs independently of traditional risk factors, such as smoking, dyslipidaemia, hypertension, etc. and is more common in those with extra- articular disease [7]. The higher incidence of IHD appears related to disease factors, such as widespread inflammation, but medications, such as NSAIDs (including selective COX-2 inhibitors) and corticosteroids, may play a role. Thus there should be a heightened awareness when ruling out ischaemic chest pain in the RA patient. Cardiac investigations are no different to those undertaken for non-RA patients and a patient with a history suggestive of acute coronary syndrome (ACS) but negative serial cardiac biomarkers and electrocardiograms must proceed to provocative testing. Management of ACS in RA is no different (see Chapter 5.2). Systemic lupus erythematosus is a multisystem, autoimmune disease. It is the prototype disease of immune complex deposition resulting in tissue damage across a wide range of organ systems and one of the most common autoimmune conditions in women of childbearing age. Common presenting features of SLE include general constitutional symptoms, such as fatigue, malaise and weight loss. There is a variety of skin manifestations in SLE which are lupus-specific (malar rash, discoid lupus, subacute cutaneous lupus erythematosus) or non-specific (panniculitis, alopecia, oral ulceration). Arthralgias or an acute non-erosive arthritis are the most common presenting symptoms of SLE. Another common manifestation is serositis causing pleurisy, pericarditis or peritonitis. SLE also causes renal and CNS disease (see below) and, rarely, can involve the lung parenchyma (pneumonitis, pulmonary hypertension) and heart (myocarditis, endocarditis). Myositis may also occur. A full blood examination often reveals cytopaenias which are a common feature of SLE. Biochemistry may indicate renal impairment. ESR and CRP may be raised. Clotting abnormalities can include a prolonged activated partial thromboplastin time due to the lupus anticoagulant (LA), one of the antiphospholipid antibodies along with anticardiolipin antibody (aCL) and others. Paradoxically, there is an associated predisposition to both venous and arterial blood clots when these are positive. The antinuclear antibody (ANA) is present in 95% of patients with SLE, but may also occur in other connective tissue and inflammatory diseases, as well as at low levels in healthy adults. The anti-Smith (Sm) and anti-dsDNA (double-stranded DNA) antibodies are more specific but less sensitive for SLE. Anti-Sm is obtained as part of a panel of antibody tests for extractable nuclear antigens (anti-ENA). Serological abnormalities also include decreased levels of complement components C3 and C4. Other tests are directed towards the organ system involved, for example, midstream urine specimen looking for proteinuria or glomerular haematuria (>70% dysmorphic red blood cells or red-cell casts) and chest X-ray in the patient with serositis. It is important to determine SLE disease activity in the ED. Useful symptoms of activity include mouth ulcers, alopecia and constitutional symptoms, as well as organ-specific symptoms, such as arthralgia or pleuritic chest pain. Investigations used to assess disease activity include complement levels (low in active SLE), CRP and ESR (elevated), as well as anti-dsDNA titre. These are not diagnostic and many people with quiescent SLE may also have hypocomplementaemia or elevated anti-dsDNA titres. A midstream urine for urinary sediment is an essential marker of renal involvement. Management of SLE is directed by the organ system involved and includes topical therapies for cutaneous lupus and NSAIDs for arthralgias and mild serositis. Most patients with SLE will be on an antimalarial, such as hydroxychloroquine, helpful for skin and musculoskeletal manifestations as well as organ involvement. Many patients will also be on corticosteroids. Those with major organ involvement will also be taking other immunosuppressants, such as methotrexate, cyclophosphamide or azathioprine. Mycophenolate mofetil is frequently now used as an alternative to cyclophosphamide for lupus nephritis. Early diagnosis of lupus nephritis is essential to prompt management and prevent progression of renal damage. Patients may be asymptomatic or present with nocturia, haematuria or proteinuria. Other presentations include hypertension, rapidly progressive glomerulonephritis and the nephrotic syndrome. Urinalysis is the most useful investigation in detecting lupus nephritis and proteinuria is the most common abnormality detected. The fresh urine specimen should be sent for phase contrast microscopy in order to detect the presence of dysmorphic erythrocytes (>70% indicates glomerular disease) or cellular casts. Urinalysis can expedite the investigation and further management of this potentially organ-threatening condition. Prompt referral to a rheumatologist or renal physician for consideration of renal biopsy and further management is indicated. There is a myriad of neuropsychiatric manifestations of neuropsychiatric SLE. Neurological presentations include: Psychiatric presentations include: These presentations are non-specific and have a broad differential diagnosis or can be subtle and progress. Unfortunately, there is no specific diagnostic test which helps differentiate SLE from other potential aetiologies. Thus, the diagnosis is made from a range of clinical features and tests. The role of the emergency department is first to exclude the more common non-SLE presentations, such as meningitis or intracranial haemorrhage. Imaging studies are needed as well as tests for SLE activity (see above). CT brain scan may detect changes of acute infarction, but is also useful in excluding other non-SLE causes, such as haemorrhage or tumour. MRI is more sensitive in detecting white matter abnormalities, although they are frequently non-specific. Cerebrospinal fluid (CSF) analysis is essential to exclude infection, but may be normal in SLE. Changes, such as elevated protein, low glucose or even a positive ANA, are non-specific and do not always reflect active SLE. The electroencephalogram is occasionally useful in cases of unexplained altered conscious level with suspected non-convulsive status epilepticus. Giant cell arteritis (GCA) is the most frequent vasculitis and almost exclusively affects Caucasians. It is a large and medium vessel vasculitis of unknown aetiology, which predominantly affects the cranial branches of arteries originating from the aortic arch and, commonly though not exclusively, the temporal artery. Polymyalgia rheumatica (PMR) is a syndrome of inflammatory pain and stiffness in the shoulder and pelvic girdles that occurs alone or frequently in association with GCA. GCA and PMR rarely ever occur before the age of 50 years [8], with a mean age at diagnosis of approximately 72 years. The incidence of GCA is roughly 1 in 500 of people over the age of 50 years, although the incidence and prevalence of PMR are less well studied. The most common symptom of GCA is new headache, usually localizing to the temporal region, although it can be more diffuse. The area is often tender and worsened by brushing the hair. Most patients complain of constitutional symptoms, such as malaise, fatigue, anorexia and weight loss. Jaw claudication (pain after a period of chewing) is the most specific symptom for GCA, although not sensitive, as it is present in only 34% [8]. On examination, the temporal arteries may be thickened, ‘ropey’ and tender with a reduced or absent pulse. The most serious complication of GCA is anterior ischaemic optic neuropathy (AION) resulting in sudden painless loss of vision which can be bilateral, particularly if untreated. Less commonly, other branches of the aorta may be involved resulting in hemiparesis, arm claudication, aortic dissection or myocardial infarction. PMR usually affects the neck, shoulder and pelvic girdles resulting in stiffness and inflammatory pain, worse in the morning and after rest. The pain is often poorly localized and muscle atrophy may occur late in the disease. There may also be synovitis affecting the shoulders, knees, wrists and hands. The relationship between onset of symptoms of GCA and PMR is highly variable. PMR symptoms may occur before, after or with GCA symptoms. At some point, 5–15% of patients with PMR will have a diagnosis of GCA and about 50% of patients with GCA have symptoms of PMR. GCA can mimic any of the other vasculitides. Non-arteritic anterior ischaemic optic neuropathy can also mimic GCA. The differential diagnosis of PMR includes late-onset RA, polmyositis and other myopathies, fibromyalgia, malignancy and hypothyroidism. The classic non-specific laboratory finding in GCA and/or PMR is a markedly elevated ESR (often>100 mm/h). CRP is also usually elevated and a full blood count often shows a mild normochromic normocytic anaemia. A temporal artery biopsy confirms the diagnosis of GCA and is particularly useful when the diagnosis is doubtful or the presentation atypical. However, as there is a false negative rate of 10–30%, a negative biopsy does not exclude GCA. The ACR classification criteria for GCA are helpful in differentiating GCA from other forms of vasculitis [9]. They include age at onset>50 years, a new headache, temporal artery tenderness or decreased pulsation and an ESR>50. An abnormal artery biopsy showing vasculitis with mononuclear infiltrate or granulomatous inflammation with multinucleated giant cells also confirms the diagnosis. Although various classification criteria for PMR have been published, having excluded other diagnoses (except GCA), the presence of all three of the following clinical and laboratory criteria defines the diagnosis [10]: In practice, rapid response to prednisolone≤20 mg daily is also used as an additional criterion with 50–70% improvement within 72 hours. Corticosteroids are essential for GCA and should not be withheld to perform a biopsy. The initial dose for GCA is unclear, but prednisone 1 mg/kg/day is used especially for ischaemic complications. However, lower doses, such as prednisone 40–60 mg, are recommended for uncomplicated disease [10]. The dose of prednisone for PMR uncomplicated by GCA is lower at 10–20 mg/day; 15 mg is generally agreed as an appropriate standard dose [11]. Most GCA patients do not require hospital admission, provided a temporal artery biopsy can be organized within a few days. However, patients with visual loss at diagnosis require urgent treatment often with pulsed parenteral corticosteroids and inpatient admission. Patients with GCA should also be commenced on aspirin. The systemic vasculitides are a group of disorders characterized by an inflammatory infiltrate in the walls of blood vessels resulting in damage to the vessel wall. The clinical manifestations depend upon the size of vessel and location in the vascular tree and may result in systemic or organ-specific manifestations. Table 14.1.2 classifies vasculitic syndromes according to vessel size (there is much overlap). Table 14.1.2 Classification of systemic vasculitis according to vessel size An underlying vasculitis should be considered in patients who present with one or more of the following: Baseline investigations should include full blood count (FBC), urea and electrolytes (U&E), liver function tests (LFTs) and clotting studies as well as CRP and ESR. Blood cultures should be taken if the patient is systemically unwell. A panel of autoimmune serological tests is carried out including ANA, ENA, rheumatoid factor, dsDNA, anticyclic citrullinated peptides (anti-CCP), complement levels (C3, C4), antineutrophil cytoplasmic antibodies (ANCA) and cryoglobulins. PAN is associated with hepatitis B and cryoglobulinaemic vasculitis with hepatitis C infection. Collection of a midstream urine specimen to look for glomerular haematuria is mandatory when vasculitis is suspected. Imaging is indicated, such as chest X-ray, CT scan of the chest or sinuses or other areas depending on the suspected organ involved. The definitive diagnosis of vasculitis requires biopsy of affected tissue or angiography. Other conditions which may mimic systemic vasculitis include: Treatment is usually with high-dose corticosteroids and, depending on the condition, additional immunosuppression, such as cyclophosphamide. Urgent specialist referral is essential. Ankylosing spondylitis (AS) is an inflammatory arthritis of the axial skeleton which can result in progressive spinal fusion. It affects<1% of the general population and its prevalence is linked to the prevalence of HLA-B27. The hallmark pathological feature of AS is new bone formation and spinal fusion which, combined with the increased prevalence of low bone mineral density in these patients, makes spinal injury a particular risk. Also a range of features including reduced mobility and muscle atrophy lead to a higher falls risk. Spinal fractures are up to four times more common in AS patients than in the general population and the risk of spinal cord injury is even higher. Fractures can occur at any point in the spine and do not have the classical appearance of wedge or endplate compression. In advanced fusion, the spine may fracture in a similar way to a long bone and not respect traditional spinal anatomical boundaries. There is also a higher rate of atlanto-axial subluxation, with similar precautions required as in those with RA. Furthermore, patients with advanced fusion may also develop cauda equina syndrome in the absence of a fracture. Fractures in AS can be missed by plain X-ray and the onset of new spinal pain or a change in spinal pain necessitiates further imaging with either CT or MRI. The medications used in rheumatology include NSAIDs, corticosteroids, DMARDs and biological DMARDs. These medications are all associated with adverse effects which occasionally result in serious morbidity. NSAIDs are commonly used for relief of arthralgia in both inflammatory and non-inflammatory conditions. They are of equal efficacy, although those with shorter half-lives appear to have less gastrointestinal toxicity [12]. NSAIDs should be used in the lowest possible dose for the shortest duration and combinations of NSAIDs (except aspirin) should be avoided [12]. The most common adverse effects of NSAIDs include peptic ulcer disease and acute renal failure, both related to inhibition of prostaglandin synthesis. Gastrointestinal (GI) toxicity is more common in the elderly, those on anticoagulants and with high doses or prolonged duration of NSAIDs. Prescribe a proton pump inhibitor when there is concern about GI toxicity. The COX-2 selective inhibitors, such as celecoxib, have a reduced incidence of peptic ulcer disease, but a similar incidence of other adverse effects including hypertension, peripheral oedema and cardiac failure. There is an increased risk of cardiovascular deaths with prolonged courses. Corticosteroids are the mainstay of treatment for most inflammatory rheumatological conditions. At high doses, they provide rapid control of inflammatory disease and are often required for long-term management at low doses. Long-term use is associated with numerous adverse effects, such as diabetes, hypertension and osteoporosis. In addition, psychosis and mood disorders related to corticosteroid use, as well as peptic ulcer disease, may present as an emergency. Although there is concern about infection among patients on DMARDs, prednisolone contributes considerably (possibly more) to the immune-suppressed patient’s overall infection risk. This heterogeneous group of medications is used to prevent joint destruction in the inflammatory arthritides and as steroid-sparing therapy in many connective tissue diseases. They include methotrexate, leflunomide, hydroxychloroquine, sulphasalazine, ciclosporin, azathioprine and cyclophosphamide. Each drug has its own range of adverse effects, but common adverse effects include cytopaenias, rashes including Stevens–Johnson syndrome, abnormal liver function tests, GI toxicity and heightened susceptibility to infections (Table 14.1.3). Table 14.1.3 Adverse effects of disease modifying antirheumatic drugs (DMARDs) The so-called ‘biological’ DMARDs are a newer and expanding collection of therapies directed against molecules and cells that mediate joint destruction and help drive the inflammatory process. These therapies are being increasingly used for those who fail conventional DMARD therapy for RA and TNF inhibitors are also used for treatment-resistant psoriatic arthritis and ankylosing spondylitis, as well as other non-rheumatological conditions. Adverse effects associated with biological DMARDs include an increased risk of infections, particularly soft-tissue and joint infections, as well as reactivation of tuberculosis. Other opportunistic infections appear more common, such as listeriosis. Patients may also develop local injection site reactions and infusion-related reactions, which can be delayed. Less common adverse effects include a form of drug-induced lupus and demyelination. As treatment of rheumatological conditions is directed at immunosuppression, infections are a common and expected adverse effect of therapy. Although studies have shown RA patients to have a de novo increased risk of infection, biological DMARDs further increase this risk. Although most of the larger studies have focused on TNF inhibitors, which have been available the longest, there is an increased risk of serious infections compared to the general RA population. This risk may be highest in the first 6 months of therapy [13]. Patients on biological therapy who develop an infection are advised temporarily to cease their treatment and to commence antibiotics. If in doubt, they should be admitted to hospital to receive parenteral antibiotics. There is also an increased risk of reactivation of tuberculosis and infections such as Listeria and Salmonella[13]. Rigorous tuberculosis screening prior to commencement of anti-TNF therapy should now be universal. Special mention must be made of the biological agent tocilizumab directed against interleukin-6. Tocilizumab causes marked suppression of acute phase reactants, particularly CRP and, even in the presence of active infection, a patient on this may have a normal CRP. Thus if there is a clinical suspicion of infection, appropriate antibiotic therapy must be instituted regardless. Anaemia, leucopaenia and thrombocytopaenia all may occur in patients taking DMARDs, such as methotrexate, cyclophosphamide, sulphasalazine and azathioprine, with neutropaenic sepsis a particular danger. Cytopaenia in a patient taking methotrexate is uncommon but those at increased risk include the elderly and those with renal impairment, related to the drug’s mechanism of action as an inhibitor of dihydrofolate reductase. Management includes temporary cessation of treatment and administration of folinic acid, the active form of folic acid which does not require to be converted by dihydrofolate reductase. The most common adverse effect of cyclophosphamide is myelosuppression, particularly leucopaenia. The white cell nadir occurs at 2 weeks post-infusion following intravenous therapy. Patients on oral therapy may experience a gradual decrease in white cell count, which is typically less predictable than on intravenous therapy. Bone marrow suppression may also occur as a side effect of azathioprine treatment, especially if given in combination with allopurinol which inhibits its metabolism, thus potentiating bone marrow toxicity. Cytopaenias are also more common in patients with deficient thiopurine methyltransferase enzyme. Sulphasalazine therapy is uncommonly complicated by bone marrow suppression. Methotrexate and leflunomide may both result in lung toxicity. The incidence of methotrexate-induced lung toxicity is difficult to assess but uncommon, with those patients at higher risk who have prolonged duration of methotrexate treatment, pre-existing rheumatoid involvement of the lungs and pleura, increased extra-articular manifestations, diabetes mellitus, previous DMARD use and a low serum albumin [14]. Age and smoking also appear to be important. The most frequent is a hypersensitivity pneumonitis, but other forms of lung injury may occur both acute and chronic, with rapid progress to respiratory failure in more acute situations. Clinical features are non-specific and include constitutional symptoms, cough and progressive dyspnoea. Subacute presentations are more common. Imaging reveals interstitial opacities and patchy consolidation. High-resolution CT scanning typically shows a ground-glass appearance. The main differential diagnosis is of a respiratory infection which may be due to typical pathogens or opportunistic infections, such as Pneumocystis jirovecii. Management is supportive with empiric antibiotic therapy in case of infection. Corticosteroids are also used. Patients may become seriously ill and require intensive care, but mortality is still low (1%). Leflunomide may also cause lung injury, typically in the first few months of therapy and usually when given in combination with methotrexate. Minor hypersensitivity reactions to allopurinol occur in about 2% of patients and usually consist of a mild rash. Rarely, a severe hypersensitivity syndrome may present in an unwell patient with fever, rash including toxic epidermal necrolysis, erythema multiforme or a diffuse macropapular or exfoliative dermatitis, abnormalities of liver function, peripheral blood eosinophilia and acute renal failure due to interstitial nephritis. It is more common in those with renal impairment who do not have an appropriate dose reduction. This presentation has a mortality rate of 25%. Treatment is supportive. Michael J Gingold, Adam B Bystrzycki and Flavia M Cicuttini The assessment of a patient with acute monoarthritis is focused on excluding a septic arthritis. Septic arthritis can cause rapid joint destruction and mortality has been reported as high as up to 15% [1]. Non-gonococcal bacterial arthritis occurs when bacteria enter the synovial lining of a joint via the haematogenous route, local spread from nearby soft-tissue infections or following penetrating trauma or injury to a joint. When the bacteria reach the synovium, they trigger an inflammatory response and bacteria and inflammatory cells enter the synovial fluid in the joint space, causing swelling and destruction of articular cartilage. These destructive changes may extend to subchondral bone and produce irreversible damage within days. The commonest causative organisms are staphylococci and streptococci. The prevalence of septic arthritis ranges between 4 and 10:100,000 patients per year and appears to be rising. It is also almost seven times more common in indigenous Australians [2]. Risk factors for septic arthritis include inflammatory arthritis (especially rheumatoid arthritis), diabetes mellitus and systemic factors, such as age greater than 80 years, as well as local factors, such as recent joint surgery, joint prosthesis and overlying skin infection. These individual risk factors increase the risk of septic arthritis by two- to threefold [3]. Skin infection overlying a prosthetic joint increases the risk of infection by 15-fold [3]. Septic arthritis presents with joint pain and swelling in over 80% of cases, which may or may not be associated with systemic symptoms, such as sweats and rigors [3]. The hip and knee joints are the most commonly involved joints. The patient may be febrile and the affected joint is usually swollen, warm, erythematous and tender. Classically, there is reduced ability to actively move the joint and marked pain on passive movement. Unfortunately, the symptoms and signs are not sensitive and a patient with septic arthritis may present with only certain of these features. Thus, septic arthritis cannot be excluded with confidence on the history and examination alone. The differential diagnosis of acute monoarthritis is shown in Table 14.2.1. Ask the patient about a history of previous rheumatological disease, such as rheumatoid arthritis, gout or other inflammatory arthritis, as well as risk factors for infection, such as immunosuppression, including diabetes and steroids. Recent trauma or history of a bleeding diathesis or anticoagulation are also relevant. Finally, ask the patient about any recent sexually transmitted infection, including gonococcal infection or non-specific urethritis, or any systemic features including uveitis and/or gastrointestinal infection, which may point towards a reactive arthritis. Table 14.2.1 Common presentations with acute monoarthritis to an emergency department Gout Reactive arthritis such as post-viral, Reiter’s syndrome Acute exacerbation of pre-existing inflammatory arthritis Rheumatoid arthritis Septic arthritis Note: Orthopaedic-related joint problems, such as trauma and/or haemarthrosis, plus osteoarthritis (OA) were not included in this series [4]. Send blood for a full blood count, which may reveal an elevated peripheral white blood cell count, as well as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). ESR and CRP are non-specific and not sensitive for septic arthritis, but may help in the differential diagnosis. Blood cultures are taken in the presence of fever. Finally, a serum urate may be elevated, but can be normal in acute gout. X-ray may be normal in septic arthritis, as it takes at least one week for destructive changes to appear on plain X-ray. Magnetic resonance imaging when available, while non-specific, is frequently helpful to determine if the pathology is in the joint or juxta-articular bone. The single most important investigation is synovial fluid aspiration and analysis. Send the aspirate in a sterile container for Gram stain and culture, as well as for polarizing light microscopy to look for the presence of urate (strongly negative birefringent) crystals or calcium pyrophosphate crystals (weakly positive birefringent crystals). Using blood culture bottles does not appear to increase the yield of a positive culture. Place some of the aspirate in an EDTA tube for a white cell count to be performed. The likelihood of septic arthritis increases from 2.9% with a synovial white cell count above 25,000/μL up to 28% with a synovial white cell count of greater than 100,000/μL. Synovial glucose and protein levels are unhelpful. There is no ‘gold standard’ test for the diagnosis of septic arthritis. Synovial fluid Gram stain has a sensitivity of up to 50% only, while culture has a sensitivity up to 85% [3]. However, combined with an appropriate clinical presentation, the presence of microorganisms in synovial fluid on Gram stain and/or a positive synovial fluid culture with high synovial white cell count are diagnostic. Treatment of septic arthritis requires parenteral antibiotics and urgent referral to orthopaedics for surgical drainage with admission to hospital. Commence empirical antibiotic therapy with dicloxacillin or flucloxacillin 2 g IV 6-hourly or cephalothin 2 g IV 6-hourly (or cephazolin) if patient is allergic to penicillin to cover against Staphylococcus, until guided by microbiology results. The patient with suspected hip sepsis or sepsis affecting a prosthetic joint must be referred to orthopaedics urgently without attempting joint aspiration. Gout is an intra-articular inflammatory response to monosodium urate crystal deposition usually related to hyperuricaemia. It is more common in males than females, but is extremely rare in the premenopausal female. Uric acid is derived from purine metabolism. Hyperuricaemia is the strongest predictor for gout and relates to either overproduction or underexcretion of uric acid. Hyperuricaemia may also cause radiolucent renal calculi. Overproduction of uric acid is due to dietary factors, such as beer, fructose-containing soft drinks, shellfish and other purine-rich foods, or endogenous factors associated with high cell turnover, such as a haematological malignancy. Reduced excretion is related to chronic kidney disease, hypovolaemia, acidosis and medications, such as diuretics, ciclosporin, pyrazinamide, ethambutol and low-dose aspirin. There is frequently a family history of gout. The peak incidence of acute gout occurs in men between the ages of 30 and 60 years and in women between 55 and 70 years. The presentation of gout in younger patients should prompt a search for a secondary cause (including lifestyle factors). Gout is more common in Maori and Polynesian populations. The classic presentation is of acute onset of a hot, swollen and painful first metatarsophalangeal joint (75% of cases) known as podagra. Other commonly affected joints include joints in the foot, the ankle, knee and small joints of the hand. Common triggers of an acute attack are binges of alcohol or purine-rich foods, dehydration, severe illness such as sepsis, trauma and surgery. Sudden cessation or the introduction (especially in an acute attack) of hypouricaemic agents, such as allopurinol or probenecid, may also precipitate gouty arthritis, as can the introduction or a dose change of a diuretic. Untreated, the symptoms will abate over the course of several days to 2 weeks. Occasionally, the patient may appear systemically unwell during an acute attack with malaise and systemic inflammatory response features. Examination reveals a tender, warm and erythematous joint with severely restricted range of movement. The patient may also be febrile. Presentations of acute gout may also be polyarticular (see Chapter 14.3). Recurrent untreated acute gout and hyperuricaemia results in chronic tophaceous gout, where the patient is no longer pain-free between attacks. Examination reveals tophus formation on the ears, around the elbows and in the fingers with marked joint deformity. Synovial fluid aspirate to identify monosodium urate crystals is diagnostic of acute gout. The crystals may be phagocytosed (intracellular) and the synovial fluid will have a high white cell count. Send fluid for Gram stain and culture to rule out septic arthritis, which may rarely coexist with gout. Podagra with a typical clinical scenario has a sensitivity of 96% and specificity of 95% for acute gout, so aspiration is not indicated [5]. Hyperuricaemia on blood testing is not diagnostic of gout as, although up to 5% of adults may have a raised serum uric acid at some point, only one-fifth (1% overall) will ever have an attack of gout. Conversely, in about one-third of patients with gout, the serum uric acid level is normal during an acute attack. Other blood tests, such as FBE, ESR and CRP are sent and may be abnormally elevated. Check the renal function with serum urea and creatinine both to identify a potential aetiology and help guide treatment, such as avoidance or reduced doses of non-steroidal anti-inflammatory drugs (NSAIDs) or colchicine. Plain X-ray is performed to exclude injury, but should be normal in the acute attack other than soft-tissue swelling. Punched-out periarticular erosions are seen in chronic gouty arthritis which, when associated with calcium deposition, deforming arthritis and soft-tissue swelling, are characteristic of chronic tophaceous gout. The aim is to treat acute pain and then prevent chronic relapse with hypouricaemic drugs. Colchicine is losing favour due to the frequency of its side effects and potential for serious toxicity. Educate all patients to correct lifestyle factors where appropriate. After excluding infection, give either an NSAID, corticosteroid and/or colchicine in the absence of contraindications. Give diclofenac 50 mg tds orally followed by 25 mg tds orally or naproxen 500 mg followed by 250 mg tds orally until symptoms subside. A selective COX-2 inhibitor, such as celecoxib 100 mg bd orally, is preferred in patients with a history of peptic ulcer disease, although there is a similar risk of renal dysfunction in the elderly or with pre-existing renal disease. Patients with gout refractory to the above treatment or in whom other treatment is contraindicated may be given corticosteroids, such as prednisolone 25–50 mg daily for 3 days, then weaned over 1–2 weeks [6,7]. An alternative approach is intra-articular corticosteroid for monoarticular gout provided sepsis has been excluded. When NSAIDs and prednisone are contraindicated, colchicine may be used. Doses of 0.5 mg 6- or 8-hourly orally have equivalent efficacy and a lower rate of gastrointestinal toxicity compared to higher doses [8]. Higher doses, such as colchicine 1.0 mg followed by 0.5 mg up to four times daily, with a maximum cumulative dose of 6–8 mg per acute attack are no longer recommended, due to increased toxicity with nausea, vomiting, diarrhoea and the risk of renal impairment. All colchicine doses should thus be less with renal impairment, as these patients are at risk of severe neuromyopathy and/or who are on statins, as this combination may increase the risk of myopathy and rhabdomyolysis. A second attack of gout usually requires urate lowering therapy, although this is not usually commenced in the emergency setting, as treatment should be delayed until the acute flare up has settled. Allopurinol, a xanthine oxidase inhibitor, prevents the production of uric acid from xanthine. It is introduced at a low dose once the acute attack has settled and gradually titrated up to a maximum of 300 mg daily [9]. Typically, the patient will remain on a low-dose NSAID (or prednisolone/low-dose colchicine) as prophylaxis against precipitating further acute attacks. Febuxostat is a new orally administered selective xanthine oxidase inhibitor for gout that may be used to reduce urate levels, particularly in patients with poor kidney function or intolerant of allopurinol, such as due to a hypersensitivity reaction. An alternative uricosuric agent to allopurinol is probenecid, which should be avoided in renal impairment. Acute pseudogout causes an acute monoarthritis and is one of the several potential presentations of calcium pyrophosphate dihydrate (CPPD) deposition disease. It is more common in females and patients over 65 years old. Calcium pyrophosphate disease is characterized by deposition of CPPD crystals in cartilage causing chondrocalcinosis. When released, there may be uptake in other synovial structures and an inflammatory response producing acute synovitis, tenosynovitis or bursitis. Advanced age is the strongest risk factor. Other associations are a family history, metabolic diseases such as haemochromatosis, Wilson’s disease, hyperparathyroidism, hypophosphataemia or hypomagnesaemia and mechanical factors, such as previous injury or osteoarthritis (OA). CPPD deposition disease presents in a variety of ways. The two most common are acute pseudogout and chronic pyrophosphate arthropathy, which may mimic OA. Other presentations include tenosynovitis, bursitis or as an incidental radiographic finding of chondrocalcinosis. CPPD deposition disease may also mimic rheumatoid arthritis or ankylosing spondylitis, as well as the neuropathic joint. Acute pseudogout typically presents in older patients and the knee is the most commonly affected joint. Other common sites include the wrist, shoulder, elbow and ankle. Occasionally, there may be an oligoarticular presentation. Presentation is with a hot, red and swollen joint. There may be systemic inflammatory response features and the patient may be febrile. Triggers include trauma, surgery or illness, but most cases are spontaneous. Diagnosis of pseudogout depends on the demonstration of CPPD crystals in synovial fluid, which is frequently blood stained. Polarizing light microscopy demonstrates weakly positive birefringent rhomboid-shaped crystals. Younger patients presenting with polyarticular chondrocalcinosis should be screened for an underlying metabolic cause, checking serum calcium, magnesium, phosphate, alkaline phosphatase, parathyroid hormone, thyroid function and iron studies. Plain X-rays of the joint may reveal chondrocalcinosis seen in fibrocartilage, such as the knee menisci, triangular cartilage of the wrist and pubic symphysis. Other characteristic findings are of marked degenerative change in joints that are not usually affected by OA. Symptoms of acute pseudogout frequently improve once the joint has been aspirated. Intra-articular injection of corticosteroid is also appropriate for acute monoarthritis, once infection has been excluded. In addition, rest and splintage for 48–72 h is beneficial. Give oral analgesics and NSAIDs similar to acute gout, particularly for polyarticular pseudogout as performing multiple joint injections is impractical and painful. Take care using NSAIDs in the elderly and use the smallest doses to avoid renal impairment and precipitating heart failure. Haemarthrosis is bleeding into a joint which may be traumatic and related to intra-articular injury or non-traumatic related to an underlying bleeding diathesis. The causes of haemarthrosis are listed in Table 14.2.2.
Rheumatology and Musculoskeletal Emergencies
14.1 Rheumatological emergencies
Introduction
Rheumatoid arthritis
Articular manifestations of rheumatoid arthritis
Acute monoarthritis
Cervical spine involvement
Imaging
Management
Extra-articular manifestations of rheumatoid arthritis
Rheumatoid vasculitis
Clinical features
Investigations
Diagnosis
Management
Other extra-articular manifestations of RA
Pulmonary disease
Cardiac disease
Sjögren’s syndrome
Felty’s syndrome
Renal disease
Neurological disease
Ischaemic heart disease in RA and other connective tissue diseases
Systemic lupus erythematosus
Clinical features
Investigations
Serological abnormalities
Assessing SLE disease activity
Management
Lupus nephritis
Neuropsychiatric SLE
Investigations
Temporal (giant cell) arteritis and other vasculitides
Temporal (giant cell) arteritis
Epidemiology
Clinical features
Polymyalgia rheumatica
Differential diagnosis of GCA and PMR
Investigations
Criteria for diagnosis
Management
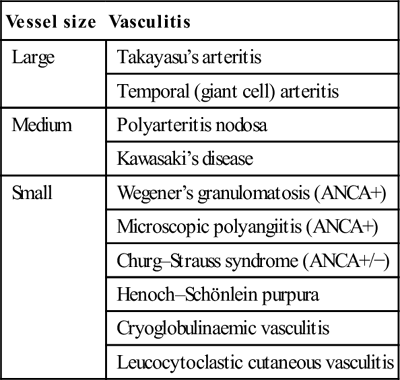
Approach to the other systemic vasculitides
Vessel size
Vasculitis
Large
Takayasu’s arteritis
Temporal (giant cell) arteritis
Medium
Polyarteritis nodosa
Kawasaki’s disease
Small
Wegener’s granulomatosis (ANCA+)
Microscopic polyangiitis (ANCA+)
Churg–Strauss syndrome (ANCA+/−)
Henoch–Schönlein purpura
Cryoglobulinaemic vasculitis
Leucocytoclastic cutaneous vasculitis
Clinical features
 unexplained systemic illness – fatigue, fevers, night sweats, malaise
unexplained systemic illness – fatigue, fevers, night sweats, malaise
 unexplained ischaemia of an organ or limb
unexplained ischaemia of an organ or limb
 chronic inflammatory sinusitis and chronic discharge or bleeding from the nose or ears
chronic inflammatory sinusitis and chronic discharge or bleeding from the nose or ears
 microscopic haematuria, especially if dysmorphic glomerular erythrocytes
microscopic haematuria, especially if dysmorphic glomerular erythrocytes
 any of the above in the setting of atopy and peripheral blood eosinophilia.
any of the above in the setting of atopy and peripheral blood eosinophilia.
Investigations and diagnosis
Differential diagnosis of systemic vasculitis
Management of systemic vasculitis
Ankylosing spondylitis
Rheumatological therapy emergencies
Non-steroidal anti-inflammatory drugs
Corticosteroids
Immunosuppressants/disease modifying antirheumatic drugs
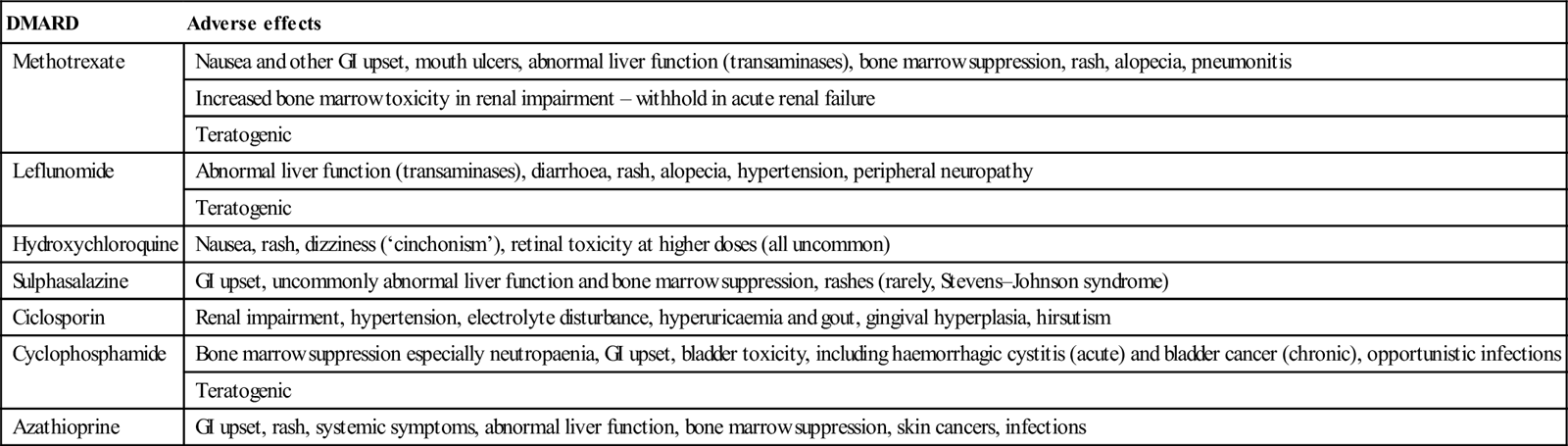
DMARD
Adverse effects
Methotrexate
Nausea and other GI upset, mouth ulcers, abnormal liver function (transaminases), bone marrow suppression, rash, alopecia, pneumonitis
Increased bone marrow toxicity in renal impairment – withhold in acute renal failure
Teratogenic
Leflunomide
Abnormal liver function (transaminases), diarrhoea, rash, alopecia, hypertension, peripheral neuropathy
Teratogenic
Hydroxychloroquine
Nausea, rash, dizziness (‘cinchonism’), retinal toxicity at higher doses (all uncommon)
Sulphasalazine
GI upset, uncommonly abnormal liver function and bone marrow suppression, rashes (rarely, Stevens–Johnson syndrome)
Ciclosporin
Renal impairment, hypertension, electrolyte disturbance, hyperuricaemia and gout, gingival hyperplasia, hirsutism
Cyclophosphamide
Bone marrow suppression especially neutropaenia, GI upset, bladder toxicity, including haemorrhagic cystitis (acute) and bladder cancer (chronic), opportunistic infections
Teratogenic
Azathioprine
GI upset, rash, systemic symptoms, abnormal liver function, bone marrow suppression, skin cancers, infections
Biological disease modifying antirheumatic drugs
Presentations of treatment-related emergencies
Infections
Bone marrow suppression
DMARD-related pneumonitis
Allopurinol hypersensitivity syndrome
14.2 Monoarthritis
Septic arthritis
Pathogenesis and pathology
Epidemiology and risk factors
Clinical features
Differential diagnosis
Clinical investigations
Blood tests
Imaging
Joint aspiration
Criteria for diagnosis of septic arthritis
Treatment
Gout
Aetiology and pathogenesis
Epidemiology
Clinical features
Investigations and diagnosis
Synovial fluid aspiration
Blood tests
Imaging
Management
Acute attack
Non-steroidal anti-inflammatory drugs
Corticosteroids
Colchicine
Recurrent attacks
Urate lowering therapy
Acute pseudogout
Aetiology and pathogenesis
Clinical features
Investigations and clinical diagnosis
Joint aspiration
Laboratory studies and imaging
Management
Haemarthrosis
Aetiology