An inherited disorder of skeletal muscle triggered in most instances by inhalation agents and/or succinylcholine, resulting in hypermetabolism, skeletal muscle damage, hyperthermia, and death if untreated
 Patients may have up to three exposures to triggering agents prior to having a reaction.
Patients may have up to three exposures to triggering agents prior to having a reaction.
Key Points
 Hypercapnia resistant to increasing minute ventilation
Hypercapnia resistant to increasing minute ventilation
 Common triggering agents—Inhalational anesthetics (sevoflurane, desflurane, halothane) and succinylcholine
Common triggering agents—Inhalational anesthetics (sevoflurane, desflurane, halothane) and succinylcholine
 Discontinue all triggering agents if Malignant Hyperthermia (MH) is suspected.
Discontinue all triggering agents if Malignant Hyperthermia (MH) is suspected.
 Control airway and ventilation
Control airway and ventilation
 Call for help and MH cart
Call for help and MH cart
 Administer dantrolene
Administer dantrolene
Common Symptoms to Remember
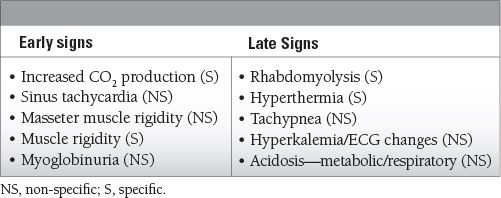
Epidemiology
 Children: 1:15,000 general anesthetics
Children: 1:15,000 general anesthetics
 Adults: 1:30,000 when general inhalational anesthetics and succinylcholine are used or 1:220,000 when general inhalation anesthetics are used only
Adults: 1:30,000 when general inhalational anesthetics and succinylcholine are used or 1:220,000 when general inhalation anesthetics are used only
Key Pathophysiology
 Patients who are susceptible to developing MH have an abnormality in their ryanodine receptor type 1.
Patients who are susceptible to developing MH have an abnormality in their ryanodine receptor type 1.
 This leads to an excessive and sometimes sustained release of calcium from the sarcoplasmic reticulum, causing persistent skeletal muscle contraction in the presence of triggering anesthetic agents.
This leads to an excessive and sometimes sustained release of calcium from the sarcoplasmic reticulum, causing persistent skeletal muscle contraction in the presence of triggering anesthetic agents.
 This contraction causes the downstream effects of rigidity, hyperthermia, excessive carbon dioxide production, and rhabdomyolysis.
This contraction causes the downstream effects of rigidity, hyperthermia, excessive carbon dioxide production, and rhabdomyolysis.
Differential Diagnosis
 Any condition that impairs elimination of carbon dioxide
Any condition that impairs elimination of carbon dioxide
 Hypoventilation
Hypoventilation
 Pneumothorax
Pneumothorax
 Bronchial obstruction
Bronchial obstruction
 Intracranial bleed
Intracranial bleed
 Traumatic brain injury
Traumatic brain injury
 Hypoxic encephalopathy
Hypoxic encephalopathy
 Thyrotoxicosis
Thyrotoxicosis
 Pheochromocytoma
Pheochromocytoma
 Sepsis
Sepsis
 Heat stroke
Heat stroke
 Serotonin syndrome
Serotonin syndrome
 Neuroleptic malignant syndrome
Neuroleptic malignant syndrome
 CO2 absorption during laparoscopy
CO2 absorption during laparoscopy
 Conditions associated with MH
Conditions associated with MH
 Central core myopathy—very high risk associated with MH
Central core myopathy—very high risk associated with MH
 King–Denborough syndrome
King–Denborough syndrome
 Multicore myopathy
Multicore myopathy
Management and Treatment
 Rising end-tidal CO2 despite increases in minute ventilation along with muscle rigidity.
Rising end-tidal CO2 despite increases in minute ventilation along with muscle rigidity.
 Exclude other causes: either decreased CO2 elimination or increased CO2 production—these should not hinder proceeding with MH treatment.
Exclude other causes: either decreased CO2 elimination or increased CO2 production—these should not hinder proceeding with MH treatment.
 Communication
Communication
 Call for MH cart and additional help
Call for MH cart and additional help
 Notify surgeon/complete operation if applicable
Notify surgeon/complete operation if applicable
 Call MH Hotline: 1-800-644-9737 (US)
Call MH Hotline: 1-800-644-9737 (US)
 Discontinue offending agents
Discontinue offending agents
 Inhalational agents and succinylcholine
Inhalational agents and succinylcholine
 100% oxygenation with controlled hyperventilation
100% oxygenation with controlled hyperventilation
 Administer dantrolene
Administer dantrolene
 Binds to ryanodine receptors and directly inhibits sarcoplasmic reticulum calcium release effectively reversing muscle hypermetabolism
Binds to ryanodine receptors and directly inhibits sarcoplasmic reticulum calcium release effectively reversing muscle hypermetabolism
 Dose: 2.5 mg/kg IV every 5 minutes up to 10 mg/kg
Dose: 2.5 mg/kg IV every 5 minutes up to 10 mg/kg
 Laboratory values
Laboratory values
 Assess electrolytes, ETCO2, blood gases, creatine phosphokinase, serum myoglobin, core temperature, urine output, and color and coagulation studies
Assess electrolytes, ETCO2, blood gases, creatine phosphokinase, serum myoglobin, core temperature, urine output, and color and coagulation studies
 Hyperkalemia
Hyperkalemia
 Treat with calcium, insulin, glucose, albuterol, bicarbonate, kayexalate, furosemide
Treat with calcium, insulin, glucose, albuterol, bicarbonate, kayexalate, furosemide
 Cardiac arrhythmias
Cardiac arrhythmias
 Resolves with treatment of acidosis and hyperkalemia
Resolves with treatment of acidosis and hyperkalemia
 Typical antiarrhythmics may be used if arrhythmias persist but calcium channel blockers are contraindicated because they can worsen hyperkalemia and result in cardiac collapse
Typical antiarrhythmics may be used if arrhythmias persist but calcium channel blockers are contraindicated because they can worsen hyperkalemia and result in cardiac collapse
 Cool the patient
Cool the patient
 Temperature goal of <38.5°C (101.3°F)—but avoid hypothermia
Temperature goal of <38.5°C (101.3°F)—but avoid hypothermia
 Continuous monitoring and support of respiratory, cardiovascular, and renal function in the intensive care unit
Continuous monitoring and support of respiratory, cardiovascular, and renal function in the intensive care unit
 Recurrence of the signs appeared in 25 patients after initial treatment; dantrolene 1 mg/kg every 6 hours should continue for 48 hours after last sign of MH resolves.
Recurrence of the signs appeared in 25 patients after initial treatment; dantrolene 1 mg/kg every 6 hours should continue for 48 hours after last sign of MH resolves.
Outcomes
 Mortality is estimated at 1% to 20%
Mortality is estimated at 1% to 20%
 Development of Disseminated Intravascular Coagulation (DIC)—greater risk for cardiac arrest and death
Development of Disseminated Intravascular Coagulation (DIC)—greater risk for cardiac arrest and death
 Susceptible individuals should avoid anesthesia with triggering agents
Susceptible individuals should avoid anesthesia with triggering agents
 Family and relatives should be informed and the susceptibility of MH should be discussed
Family and relatives should be informed and the susceptibility of MH should be discussed
SUGGESTED READINGS
Burkman JM, Posner KL, Domino KB. Analysis of the clinical variables associated with recrudescence after malignant hyperthermia reactions. Anesthesiology. 2007;106:901.
D’Arcy CE, Bjorksten A, Yiu EM, et al. King-Denborough syndrome caused by a novel mutation in the ryanodine receptor gene. Neurology. 2008;71:776.
Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131.
Guis S, Figarella-Branger D, Monnier N, et al. Multiminicore disease in a family susceptible to malignant hyperthermia: histology, in vitro contracture tests, and genetic characterization. Arch Neurol. 2004;61:106.
Larach MG, Brandon BW, Allen GC, et al. Cardiac arrests and deaths associated with malignant hyperthermia in North America from 1987 to 2006: a report from the North American Malignant Hyperthermia Registry of the Malignant Hyperthermia Association of the United States. Anesthesiology. 2008;108:603.
Larach MG, Gronert GA, Allen GC, et al. Clinical presentation, treatment, and complications of malignant hyperthermia in North America from 1987 to 2006. Anesth Analg. 2010;110:498.
Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771.
Malignant Hyperthermia Association of the United States. http://www.mhaus.org/
Rosenberg H, Davis M, James D, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007; 2:21.
Rosero EB, Adesanya AO, Timaran CH, et al. Trends and outcomes of malignant hyperthermia in the United States., 2000 to 2005. Anesthesiology. 2009; 110:89.
Zhang Y, Chen HS, Khanna VK, et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet. 1993;5:46.
13.2
Neuroleptic Malignant Syndrome
Shamim Nejad
Neuroleptic Malignant Syndrome
 A very rare but potentially fatal syndrome associated with the use of dopamine antagonist medications (antiemetics, antipsychotics, etc.) commonly used in critical care medicine.
A very rare but potentially fatal syndrome associated with the use of dopamine antagonist medications (antiemetics, antipsychotics, etc.) commonly used in critical care medicine.
 Characterized by administration of D2-blocking drugs and early development of hyperthermia, muscle rigidity, mental status changes, and autonomic instability (hyper- or hypotension, tachycardia, tachypnea, and diaphoresis). Other drug-induced, systemic, or other neuropsychiatric illnesses should be excluded before making a diagnosis of neuroleptic malignant syndrome (NMS).
Characterized by administration of D2-blocking drugs and early development of hyperthermia, muscle rigidity, mental status changes, and autonomic instability (hyper- or hypotension, tachycardia, tachypnea, and diaphoresis). Other drug-induced, systemic, or other neuropsychiatric illnesses should be excluded before making a diagnosis of neuroleptic malignant syndrome (NMS).
 Risk Factors
Risk Factors
 History of NMS or catatonia
History of NMS or catatonia
 Pre-existing organic brain disease, especially involving diminished central brain dopamine activity or receptor function (e.g., Parkinson’s disease, traumatic brain injury)
Pre-existing organic brain disease, especially involving diminished central brain dopamine activity or receptor function (e.g., Parkinson’s disease, traumatic brain injury)
 Use of high-potency antipsychotics
Use of high-potency antipsychotics
 Dehydration
Dehydration
 Patients with significant fluid shifts (i.e., postpartum women)
Patients with significant fluid shifts (i.e., postpartum women)
 Agitated men under the age of 40 years old
Agitated men under the age of 40 years old
 Genetic susceptibility (reports of incidence amongst certain families)
Genetic susceptibility (reports of incidence amongst certain families)
 Clinical characteristics: NMS presents as a classic tetrad in the setting of neuroleptic use
Clinical characteristics: NMS presents as a classic tetrad in the setting of neuroleptic use
 fever
fever
 rigidity (lead-pipe)
rigidity (lead-pipe)
 autonomic instability
autonomic instability
 altered mental status
altered mental status
 Early signs include unexpected mental status changes and muscle rigidity developing over the course of a few days (range: hours to weeks) in association with the introduction of D2 blockade or removal of dopamine potentiation.
Early signs include unexpected mental status changes and muscle rigidity developing over the course of a few days (range: hours to weeks) in association with the introduction of D2 blockade or removal of dopamine potentiation.
 Mental status changes are nonspecific and typically include confusion and clouding of consciousness, with approximately 50% having abnormal EEGs. Hyperthermia usually develops late. Autonomic dysfunction may develop at any point.
Mental status changes are nonspecific and typically include confusion and clouding of consciousness, with approximately 50% having abnormal EEGs. Hyperthermia usually develops late. Autonomic dysfunction may develop at any point.
 Laboratory abnormalities
Laboratory abnormalities
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


