Edited by Mark Little David McCoubrie and Alan Gault Acid–base disorders are commonly encountered in the emergency department (ED) and their recognition is important for the diagnosis, assessment of severity and monitoring of many disease processes. Although these disorders are usually classified according to the major metabolic abnormality present (acidosis or alkalosis) and its origin (metabolic or respiratory), it is important to realize that acid–base disorders of a mixed type commonly occur and that the recognition and assessment of these are more complex. CO2 produces acid when in solution and altering PaCO2 through changes in ventilation can produce or remove acid from the body. The terms respiratory acidosis/alkalosis refer to the pH shifts resulting from alterations in PaCO2 from changes in ventilation. Bicarbonate acts as a base in solution with bicarbonate accumulation resulting in a more alkaline state and its wasting or consumption indicating a more acidic state. The terms metabolic acidosis/alkalosis refer to pH shifts characterized by alterations in bicarbonate levels. By convention, the overall pH abnormality as defined by the blood gas assessment is termed alkalaemia (for pH>7.44) or acidaemia (pH<7.34). Acid–base status is one of the most tightly regulated systems in the body. The term compensation is used to describe the processes by which shifts in plasma pH are attenuated. These mechanisms include buffering, respiratory manipulation of CO2 and renal handling of bicarbonate. Buffering with plasma proteins, haemoglobin and the carbonic-acid–bicarbonate systems provide the most immediate mechanism. This is followed by respiratory compensation, which occurs within minutes and is achieved by alterations in alveolar ventilation. Renal compensation usually takes hours to days to take effect. Systemic acidaemia is defined as the presence of an increased concentration of H+ ions in the blood. An acidaemia can result from respiratory acidosis, metabolic acidosis or both. The physiological effects of acidaemia are a decrease in the affinity of haemoglobin for oxygen and an increase in serum K+ of approximately 0.4–0.6 mmol/L for each decrease in pH of 0.1 [1]. Although the presence of acidaemia is often associated with a poor prognosis, the presence of acidaemia per se usually has few clinically significant effects. It is the nature and severity of the underlying illness that principally determines outcome. A decrease in measured serum HCO3− of up to 5 mmol/L has also been reported as a result of underfilling of vacuum-type specimen tubes [2]. Metabolic acidosis is defined as an increase in the [H+] of the blood as a result of increased acid production or bicarbonate wasting from the gastrointestinal (GI) or renal tract. The cause is often multifactorial and can be further classified into ‘anion-gap’ and ‘non-anion-gap’ (or hyperchloraemic) metabolic acidosis. As electroneutrality must exist in all solutions, the anion gap represents the concentration of anions that are not commonly measured. The most commonly used formula for the calculation of the anion gap is: The normal value for the anion gap depends on the type of biochemical analyser used and, while the upper limit of normal has been commonly quoted as 14, the mean range with some modern analysers is only 5–12 [3]. In the normal resting state, the serum ionic proteins account for most of the anion gap, with a lesser contribution from other ‘unmeasured’ anions, such as PO4− and SO4−. In pathological conditions where there is an increase in the concentration of unmeasured anions, an AGMA results. The anions responsible for the increase in the anion gap depend on the cause of the acidosis. Lactic acid is the predominant anion in hypoxia and shock, PO4− and SO4− in renal failure, ketoacids in diabetic and alcoholic ketoacidosis, oxalic acid in ethylene glycol poisoning and formic acid in methanol poisoning. Of the causes of an AGMA, lactic acidosis is the most commonly encountered in the ED and is defined as a serum lactate of>2.5 mmol/L (Table 12.1.1). The presence of lactic acidosis is determined by the balance between lactate production and metabolism. In the seriously ill patient, it is common for increased production and decreased metabolism to be present simultaneously. It is important to realize that, in many conditions, a variety of factors may produce the acidosis and that multiple anions may be involved in the production of an anion-gap acidosis. In a patient with an AGMA, a non-anion-gap metabolic acidosis may also exist (see below). Non-anion-gap metabolic acidosis results from loss of HCO3− from the body, rather than increased acid production. To maintain electroneutrality, chloride is usually retained by the renal tubules when HCO3− is lost and the hallmark of non-anion-gap acidosis is an elevation of the serum chloride. The causes of non-anion-gap metabolic acidosis are further classified according to the site of HCO3− loss. Gastrointestinal losses can occur with lower gastrointestinal tract (GIT) fluid losses that are rich in HCO3− or with cholestyramine ingestion due to binding of HCO3− in the gut. Renal losses can occur with renal tubular acidosis, carbonic anhydrase inhibitor therapy or adrenocortical insufficiency. Occasionally, direct chloride excess drives the renal bicarbonate loss (again due to electroneutrality) – which can be observed with large volume chloride rich crystalloid administration (chiefly normal saline). Renal tubular acidosis (RTA) is a group of conditions where there is an impaired ability to secrete H+ in the distal convoluted tubule or absorb HCO3− in the proximal convoluted tubule. This may result in a chronic metabolic acidosis, with hypokalaemia, nephrocalcinosis, rickets or osteomalacia. There are many subtypes of RTA and many different causes. Most commonly, it is observed in patients with chronic renal impairment but it may also be drug induced with agents such as ibuprofen, toluene and carbonic anhydrase inhibitors most often implicated. The treatment of acidosis should usually be directed primarily towards correction of the underlying cause. Intravenous HCO3− is of use in the presence of acidosis and severe hyperkalaemia, severe sodium channel (e.g. tricyclic antidepressant), salicylate and methanol poisoning. The use of HCO3− in patients with diabetic ketoacidosis and lactic acidosis associated with sepsis or severe cardiorespiratory disease does not appear to improve outcome [4–6]. The potential hazards of HCO3− therapy include a high solute load, hyperosmolarity, hypokalaemia, decreased ionized serum calcium and worsening of intracellular and cerebrospinal fluid acidosis (which may precipitate hepatic encephalopathy in susceptible patients). Respiratory acidosis is defined as an elevation of the arterial partial pressure of carbon dioxide (PCO2) and is due to alveolar hypoventilation. This can result from central depression in respiriatory drive, neuromuscular weakness, mechanical factors, lung parenchymal disorders and ventilation/perfusion mismatch. With significant elevations in CO2, sweating, tachycardia, confusion and mydriasis occur. When the PCO2 is greater than 80 mmHg, the level of consciousness is usually depressed, known as CO2 narcosis. The treatment of respiratory acidosis is directed towards reversal of the causative factors while supporting and promoting ventilation. Indications for and methods of therapy are clinically determined. Alkalaemia is defined as a decrease in [H+] in the blood. Extreme alkalaemia may cause altered mental status, tetany and seizures. These are predominantly related to a reduction in the concentration of ionized calcium, which is more commonly present in respiratory alkalosis due to anxiety, than from other causes. Alkalaemia in patients with chronic airways disease may exacerbate tissue hypoxia due to leftward shift of the oxygen-dissociation curve. Like acidaemia, there are metabolic and respiratory processes by which it occurs. Metabolic alkalosis most commonly results from loss of acid from the GIT, however, renal acid losses or accumulation of bicarbonate from exogenous sources can contribute. Diagnostically and therapeutically, metabolic alkalosis can be divided into two distinct aetiological groups – chloride-responsive and chloride-unresponsive metabolic alkalosis (Table 12.1.2). Table 12.1.2 Low urine chloride variety (saline-responsive) Gastric volume loss (vomiting, nasogastric suction, bulimia nervosa) Diuretics Licorice Hypokalaemia High urine chloride variety (not saline-responsive) Primary and secondary hyperaldosteronism Apparent mineralocorticoid excess Liddle’s syndrome Conn’s syndrome (aldosteronoma) Cushing’s disease Bartter’s syndrome Gitelman’s syndrome Excess bicarbonate administration – antacids, dialysis, milk-alkali syndrome From Murray L, Daly F, Little M, Cadogan M. Toxicology Handbook, 2nd edn. Churchill Livingstone; 2011. Chloride-responsive metabolic alkalosis arises from conditions that result in both chloride and volume loss. Reduction in extracellular volume leads to increased mineralocorticoid activity causing reabsorption of sodium and secretion of hydrogen. This, in turn, causes increased formation of bicarbonate which, ultimately, overwhelms the kidneys’ ability further to excrete it. In alkalosis, the urine is usually alkaline with higher concentrations of bicarbonate, there is minimal chloride excreted in order to maintain electroneutrality. Hence a urinary chloride<10 mmol/L is a common finding in these conditions. The commonest causes seen in the emergency department are upper GIT losses as a result of severe and prolonged vomiting and diuretic use. Chloride-unresponsive metabolic alkalosis is typically due to disease states that either result in mineralocorticoid excess in the absence of hypovolaemia or chloride wasting, or congenital disorders with defects in the various ionic transport channels within the kidney. As extracellular volume is either normal or increased, urinary chloride is typically>10 mmol/L. These conditions are seen in the emergency department infrequently. Treatment should be directed primarily towards correction of the underlying cause. In the presence of upper gastrointestinal fluid losses, intravenous fluids with high chloride content (such as 0.9% saline) should be used initially for rehydration and correction of hypokalaemia is also required. Respiratory alkalosis may be acute or chronic, of which the acute form is most commonly encountered in the emergency department. Respiratory alkalosis may physiologically occur in the general population secondary to exercise, altitude-related hypoxia and stimulation of the medullary respiratory centre by progesterones during pregnancy. Disease states that give rise to respiratory alkalosis are more likely to be seen in the emergency department (Table 12.1.3). Treatment is again directed towards correction of the underlying cause. Table 12.1.3 Causes of respiratory alkalosis A systematic stepwise approach to acid–base interpretation is beneficial in the evaluation of disturbances as they are often multiple. What follows is an example of a conventional methodology as outlined by Whittier and Rutecki [7]: In an anion-gap metabolic acidosis, this step determines whether there is a non-anion-gap (hyperchloraemic) component as a contributing explanantion of the bicarbonate fall. There should be a 1:1 relationship between the rise in the anion gap over normal and the decrease in the bicarbonate. If the bicarbonate is higher than predicted then a metabolic alkosis is also present. If the bicarbonate is lower than predicted then a non-anion-gap acidosis is also present. John Pasco Hyponatraemia, defined as serum sodium concentration of less than 130 mmol/L, is a common condition. The prevalence is estimated at 2.5% in hospitalized patients, of which two-thirds develop the condition while in hospital. Hyponatraemia is almost always associated with extracellular hypotonicity, with an excess of total body water relative to sodium (hypotonic hyponatraemia). The exceptions are:
Metabolic Emergencies
12.1 Acid–base disorders
Introduction
Acid–base homeostasis
Acidaemia
Metabolic acidosis
Anion-gap metabolic acidosis (AGMA)
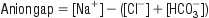
Non-anion-gap metabolic acidosis
Renal tubular acidosis
Treatment of metabolic acidosis
Respiratory acidosis
Treatment
Alkalaemia
Metabolic alkalosis
Respiratory alkalosis
CNS-mediated hyperventilation
Pulmonary
Increased intracranial pressure
Congestive cardiac failure
Cerebrovascular accidents
Mechanical hyperventilation
Psychogenic
Pneumonia
Pulmonary emboli
Hypoxia-mediated hyperventilation
Sepsis
Altitude
Toxin-induced hyperventilation
Anaemia
Nicotine
V/Q mismatch
Salicylate
Xanthines
Systematic acid–base interpretation
Step 1: what is the pH (primary acid–base disturbance)?
Step 2: determine whether the primary process is respiratory, metabolic or both
Step 3: calculate the anion gap
Causes of anion-gap acidosis (mnemonic ‘CAT MUDPILES’)
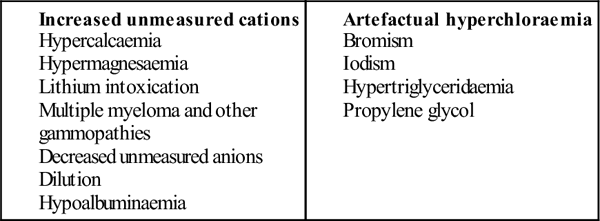
Causes of a low anion gap (<6)
Causes of non-anion-gap metabolic acidosis
Causes of respiratory acidosis
Acute
Chronic
Airway obstruction
Lung diseases, e.g. COPD, pulmonary fibrosis
Aspiration
Neuromuscular disorders, e.g. muscular atrophy
Bronchospasm
Obesity
Drug-induced CNS depression
Severe kyphoscoliosis
Hypoventilation of CNS or muscular origin
Hypoventilation of PNS origin, e.g. GBS, OP poisoning
Pulmonary disease
Step 4: check for the degree of compensation
Step 5: determine if there is a 1:1 relationship between the anions in the blood (presence of a delta gap)
12.2 Electrolyte disturbances
Hyponatraemia
Introduction
Pathophysiology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


12. Metabolic Emergencies

Only gold members can continue reading. Log In or Register to continue
