Edited by Anthony Brown Anthony Brown The classification system and diagnostic criteria for diabetes were revised in 2010 by the American Diabetes Association [1]. The classification of type I and type II diabetes mellitus was retained, with the recommended criteria for the diagnosis of diabetes as a fasting plasma glucose of 7 mmol/L or greater, or a random plasma glucose of over 11 mmol/L associated with polyuria, polydipsia and weight loss. In addition, an HbA1C (glycated haemoglobin) level of≥6.5% was added. The oral glucose tolerance test is no longer routinely recommended. The exact aetiology of diabetes is unclear. Type I diabetes is characterized by pancreatic beta cell destruction with an absolute insulin deficiency usually, but not exclusively, associated with autoimmune damage from a range of antibodies including to islet cells (ICA), glutamic acid decarboxylase (GAD), insulin and tyrosine phosphatases. Genetic and environmental factors are implicated, such as some human leucocyte antigen (HLA) types (most Caucasian patients are HLA-DR3 or DR4 or both), and abnormal immune responses, such as following viral infection. Certain genes are also implicated as co-contributors, particularly sites on chromosomes 6, 7, 11, 14 and 18. Type II diabetes results from a progressive insulin secretory deficiency on the background of insulin resistance [1]. Genetic factors are implied by strong familial aggregation of cases and environmental factors in the context of genetic susceptibility, including obesity and diet. Thus, the introduction of a high fat and high calorie ‘Western’ diet rather than traditional crop foods has seen countries such as India record among the fastest growth rate of new diabetes worldwide. Although type I diabetes occurs most frequently among Caucasians throughout the world, diabetes in Australia is three times more common in the Aboriginal community. Other groups with a high prevalence include Pacific Islanders and Native Americans. Diabetes mellitus may be secondary to conditions that damage the exocrine pancreas including chronic pancreatitis, carcinoma of the pancreas and pancreatectomy, haemochromatosis, cystic fibrosis, pregnancy (gestational) and endocrinopathies, such as Cushing’s syndrome, acromegaly, phaeochromocytoma and glucagonoma [2]. Certain drugs can impair glucose tolerance or cause overt diabetes mellitus. These include glucocorticoids, the oral contraceptive pill, thiazide diuretics at higher doses, tacrolimus, sirolimus and ciclosporin, pentamadine (which may also cause hypoglycaemia) and HIV protease inhibitors. Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic state (HHS) are both life-threatening acute complications of diabetes mellitus. Although important differences do exist, the pathophysiology and treatment are similar. DKA is usually seen in type I diabetes and HHS in patients with type II, but both complications can occur in type I and type II diabetes. See Chapter 11.2 for the diagnosis and management of DKA and HHS. The aim of optimal long-term blood sugar control is an HbA1c (glycated haemoglobin) level of less than 7.5% without frequent disabling hypoglycaemia for the prevention of microvascular disease, and 6.5% in those at increased risk of arterial disease [3]. This should be represented by a pre-prandial blood glucose level of 4.0–7.0 mmol/L and a post-prandial blood glucose level of less than 9.0 mmol/L. Insulin was first administered to humans in 1922. Animal insulins (bovine, porcine) have been used for many years but, in the 1980s, human insulins became commercially available. Today, with the widespread availability of human insulins, animal insulins are of historical interest only. Table 11.1.1 lists the different types of insulins and the important parameters of each type. Mixtures of short- and intermediate-acting insulins are also available: 70/30 (70% NPH/30% regular) and 50/50 (50% NPH/50% regular). Table 11.1.1 Pharmacokinetic characteristics of currently available human insulins Two major groups of oral hypoglycaemic agents used in the management of type II diabetes are the sulphonylureas and the biguanides. The sulphonylurea group of drugs acts by stimulating the pancreatic secretion of insulin, and the biguanide metformin acts by suppressing hepatic glucose production and enhancing the peripheral use of glucose. It is the first choice medication particularly in the overweight patient. Newer types of oral agents now available for the treatment of diabetes include the alpha-glucosidase inhibitor acarbose, which acts on the gastrointestinal tract to interfere with carbohydrate digestion, but flatulence and diarrhea may be troublesome. The thiazolidinediones, such as pioglitazone and rosiglitazone, act primarily by reducing insulin resistance, thereby enhancing the effect of circulating insulin. Roziglitazone increases the risk of myocardial infarction and cardiovascular deaths and thus should be avoided in ischaemic heart disease [4]. All thiazolidinediones must be avoided in people with moderate or severe heart failure. Finally, the dipeptidyl peptidase 4 (DDP-4) inhibitors, such as sitagliptin, inhibit DDP-4 to prolong the action of the incretin hormones. Angiotensin-converting enzyme inhibitors delay the onset of diabetic nephropathy even in normotensive patients with diabetes. Statins are important in the strict treatment of dyslipidaemia in diabetic patients. Hypoglycaemia is more common in type I diabetes. The critical plasma level at which hypoglycaemia manifests varies between different individuals, but symptoms are likely below a plasma glucose of 3.5 mmol/L. Precipitants include exercise, a late meal, inadequate carbohydrate intake, errors of insulin dosage and ethanol ingestion. Hypoglycaemia may also occur in the non-diabetic patient, precipitated by a variety of conditions (Table 11.1.2) [5]. Table 11.1.2 Hypoglycaemia produces neurological and mental dysfunction from tremor, sweating and anxiety to seizures and coma. Less commonly, it can present as hypothermia, depression and psychosis. In some instances, hypoglycaemia is relatively asymptomatic. Anthony Brown and Richard D Hardern Essentials 3 Key management components of DKA include: 4 Meticulous monitoring and documentation of treatment in DKA are essential. 7 Treatment of HHS is similar to DKA except: Diabetic ketoacidosis (DKA) is an acute, potentially life-threatening complication in an insulin-dependent diabetic and in some type II diabetics. It consists of the triad of ketonaemia, hyperglycaemia and acidaemia – a high anion-gap metabolic acidosis. Although DKA is preventable, its prevalence and suboptimal management may highlight shortfalls in the quality of care for patients with diabetes. The mortality rate in developed countries has dropped to<1%. Hyperosmolar hyperglycaemic state (HHS) is characterized by hypovolaemia, marked hyperglycaemia (>30 mmol/L) without ketonaemia or acidosis and a raised osmolality usually>320 mosmol/kg. It comes on more insidiously and has a worse prognosis with an increased mortality of around 15–20% and greater morbidity, in part related to underlying chronic medical disorders. An annual incidence of DKA of approximately 1:170 patients with type I diabetes is reported, or 2 episodes per 100 patient years of diabetes, for a prevalence of 4.6–8 episodes per 1000 patients with diabetes [1,2]. DKA may be the presenting feature of diabetes mellitus (3–25% DKA), but is usually seen in patients with autoimmune type I diabetes when there has been an insulin error with inadequate insulin or poor compliance (30% DKA), and/or an intercurrent illness (35% DKA). A variant of type II diabetes is also ketosis prone, ‘ketosis-prone (type II) diabetes’, usually in the obese with a strong family history. This was originally described in Africans and African-Americans, but is now noted worldwide. DKA arises from an absolute or relative lack of insulin accompanied by an increase in counter-regulatory hormones, such as glucagon, cortisol and growth hormone [1,3]. Insulin absence leads to increased hepatic gluconeogenesis and glycogenolysis, with an incomplete lack of insulin related to greater hyperosmolarity (in HHS). Lack of insulin and excess counter-regulatory hormones increase lipolysis and free fatty acid production as an alternate energy source. This leads to subsequent ketone body formation produced from acetyl coenzyme A, mainly in hepatic mitochondria, including acetone, beta-hydroxybutyrate and acetoacetate and a reduced ability to prevent ketonaemia. HHS is the other end of the hyperglycaemia spectrum from DKA, occurring with a relative rather than an absolute deficiency of insulin leading to a greater level of hyperglycaemia, therefore higher hyperosmolarity than is seen in DKA. The degree of dehydration is greater (typically 10–15% body weight), but significant ketosis does not occur. HHS is more insidious in onset than DKA and patients with HHS are typically older with pre-existing type II diabetes. However, HHS is seen in young adults and even teenagers. Malaise and fatigue on a background of polyuria, polydipsia, weakness and fatigability are common, but gastrointestinal symptoms, such as nausea, vomiting and abdominal pain, may predominate [3]. A lack of history of diabetes does not rule out the diagnosis of DKA, as it may be a first presentation, often presaged by recent, unexplained rapid weight loss. Laboured, sighing respirations with an increased rate and depth, known as Kussmaul breathing, are characteristic of DKA in association with dehydration causing decreased tissue turgor, a dry mouth and sweet foetor of pear drops (ketotic), which is not always noticed. The conscious level may be reduced, but coma is rare. Look carefully for signs of an underlying precipitating cause including chest, urine or skin infection, such as boils, as well as meningitis or an acute abdomen, although non-surgical upper abdominal pain is common in DKA. In those with HHS, look out for the complications of acute myocardial infarction, stroke or arterial thrombosis [4,5]. The urine output should be measured regularly, which does not always require urinary catheterization. Likewise, invasive haemodynamic monitoring should not be instituted as a ‘routine’ for patients with DKA or HHS. It should be reserved for those who fail to respond or in the elderly who are at risk of fluid overload. In addition, venous blood gases are sufficient to monitor progress in DKA, rather than repeated arterial sampling. Note that there is no precise definition of HHS, but it is characterized by [5]: Measure capillary blood glucose hourly until near to the normal range, or measure venous urea and electrolytes (U&Es), glucose and pH initially hourly, then 2-hourly once the venous glucose and capillary glucose are in agreement. A mild leucocytosis is common in DKA, which should not be interpreted as signifying infection. Likewise hyperamylasaemia is common and does not imply pancreatitis. DKA is highly likely in the presence of glycosuria and ketonuria in an unwell patient. Perform an early ECG to look for T-wave changes as a first indicator of hyperkalaemia (tall and peaked) or hypokalaemia (flat or inverted), or a clinically silent myocardial infarction (Ml) as a precipitant of HHS or DKA. Point-of-care testing for blood ketones, such as beta-hydroxybutyrate, is helpful at triage and when monitoring the response to treatment. Other causes of high (greater than 16) anion-gap metabolic acidosis include: Other causes of hyperglycaemia include: The treatment of DKA is rigorous and requires careful monitoring of the patient, both clinically and biochemically. Ideally, all observations and results are entered onto a purpose-designed record sheet, such as an integrated care pathway that includes data recording and guidance [7]. Severe DKA necessitating intensive management includes a venous or arterial pH<7.1, bicarbonate<5 mmol/L, hypokalaemia on admission (<3.5 mmol/L), Glasgow coma scale (GCS)<12, systolic BP<90 mmHg and SaO2<92% on room air. Intravenous fluids should be started within 30 min of the patient’s arrival in the ED. A shocked patient should receive a fluid bolus on arrival to restore perfusion, although care is needed in patients with co-morbidities, such as the elderly with heart or renal impairment, to avoid fluid overload. Give patients who are not shocked 1 L 0.9% normal saline over 1 hour, then at a rate of 500 mL/h for 4 h with added potassium, then 250 mL/h for the next 8 h, again with added potassium [1]. A suggested fluid regimen is shown in Table 11.2.1. Table 11.2.1
Endocrine Emergencies
11.1 Diabetes mellitus and hypoglycaemia: an overview
Diabetes mellitus
Classification system and diagnostic criteria
Aetiology
Diabetes secondary to other conditions
Drug-induced diabetic state
Emergency presentations of a high blood sugar
General management of diabetes mellitus
Aims of long-term blood sugar control
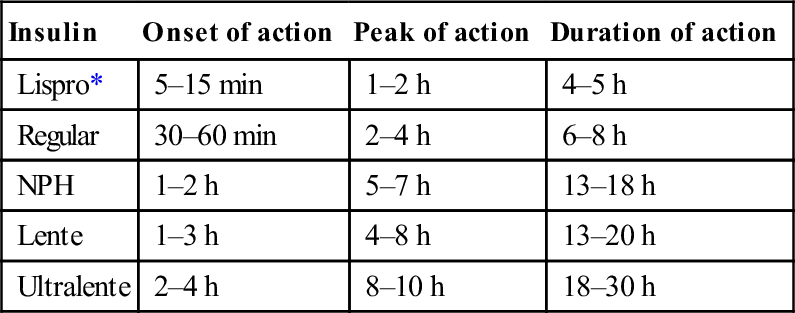
Insulins
Types of insulins
Insulin
Onset of action
Peak of action
Duration of action
Lispro*
5–15 min
1–2 h
4–5 h
Regular
30–60 min
2–4 h
6–8 h
NPH
1–2 h
5–7 h
13–18 h
Lente
1–3 h
4–8 h
13–20 h
Ultralente
2–4 h
8–10 h
18–30 h
Antidiabetic drugs
Other non-diabetic drugs
Diabetic hypoglycaemia
Diabetic patients

Clinical features
Management of hypoglycaemic coma
 The ABC approach is important in the patient with coma.
The ABC approach is important in the patient with coma.
11.2 Diabetic ketoacidosis and hyperosmolar, hyperglycaemic state
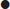 fluids (0.9% normal saline) to replace deficits of sodium of 7–10 mmol/kg and water 100 mL/kg
fluids (0.9% normal saline) to replace deficits of sodium of 7–10 mmol/kg and water 100 mL/kg
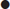 lower dose insulin infusion rate is used at 0.05 unit/kg/h
lower dose insulin infusion rate is used at 0.05 unit/kg/h
 this infusion rate is titrated against the serum osmolarity rather than ketoacids
this infusion rate is titrated against the serum osmolarity rather than ketoacids
 low molecular weight heparin (LMWH) thromboprophylaxis is indicated.
low molecular weight heparin (LMWH) thromboprophylaxis is indicated.
Introduction
Epidemiology, aetiology and pathogenesis
Pathogenesis
Clinical features
Diagnostic criteria
DKA
HHS
Typical deficits per body weight
DKA
HHS
Investigations
Venous blood
Urinalysis
Electrocardiograph (ECG)
Point-of-care testing
Differential diagnosis of DKA
 alcoholic or starvation ketoacidosis
alcoholic or starvation ketoacidosis
 lactic acidosis (multiple causes)
lactic acidosis (multiple causes)
 methanol, ethylene glycol (note ethanol predominantly leads to a lactic acidosis)
methanol, ethylene glycol (note ethanol predominantly leads to a lactic acidosis)
 drugs, such as corticosteroids, octreotide, thiazide diuretics, ritonavir, diazoxide and atypical antipsychotics (clozapine, olanzapine, risperidone, quetiapine – these may actually precipitate DKA soon after commencement, even in the absence of weight gain) [6]
drugs, such as corticosteroids, octreotide, thiazide diuretics, ritonavir, diazoxide and atypical antipsychotics (clozapine, olanzapine, risperidone, quetiapine – these may actually precipitate DKA soon after commencement, even in the absence of weight gain) [6]
 endocrine, such as Cushing’s, acromegaly, phaeochromocytoma, glucagonoma, VIPoma.
endocrine, such as Cushing’s, acromegaly, phaeochromocytoma, glucagonoma, VIPoma.
Management
Diabetic ketoacidosis
Severe DKA
Fluid regimen
Fluid rate
Litre
Time (hours from starting treatment)
First at 1000 mL/h
0–1
Second at 500 mL/h+K
1–3
Third at 500 mL/h+K
3–5
Fourth at 250 mL/h+K
5–9
Fifth at 250 mL/h+K
9–13
Reassess cardiovascular status at 12 h and adjust rate accordingly
Sixth at 166 mL/h+K
13–19
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


11. Endocrine Emergencies
Only gold members can continue reading. Log In or Register to continue
