Fig. 18.1
Putative mechanism of endocannabinoid-mediated retrograde signaling in the nervous system. Activation of metabotropic glutamate receptors (mGluR) by glutamate triggers the activation of the phospholipase C (PLC)-diacylglycerol lipase (DGL) pathway to generate the endocannabinoid 2-arachidonoylglycerol (2-AG). First, the 2-AG precursor diacylglycerol (DAG) is formed from PLC-mediated hydrolysis of membrane phospholipid precursors (PIPx). DAG is then hydrolyzed by the enzyme DGL-α to generate 2-AG. 2-AG is released from the postsynaptic neuron and acts as a retrograde signaling molecule. Endocannabinoids activate presynaptic CB1 receptors which reside on terminals of glutamatergic and GABAergic neurons. Activation of CB1 by 2-AG, anandamide, or exogenous cannabinoids (e.g., tetrahydrocannabinol, THC) inhibits calcium influx in the presynaptic terminal, thereby inhibiting release of the primary neurotransmitter (i.e., glutamate or GABA) from the synaptic vesicle. Endocannabinoids are then rapidly deactivated by transport into cells (via a putative endocannabinoid transporter) followed by intracellular hydrolysis. 2-AG is metabolized by the enzyme monoacylglycerol lipase (MGL), whereas anandamide is metabolized by a distinct enzyme, fatty-acid amide hydrolase (FAAH). Note that MGL co-localizes with CB1 in the presynaptic terminal, whereas FAAH is localized to postsynaptic sites. The existence of an endocannabinoid transporter remains controversial. Pharmacological inhibitors of either endocannabinoid deactivation (e.g., FAAH and MGL inhibitors) or transport (i.e., uptake inhibitors) have been developed to exploit the therapeutic potential of the endocannabinoid signaling system in the treatment of pain (Figure by authors with kind assistance of James Brodie, GW Pharmaceuticals)
A Brief Scientific History of Cannabis and Pain
Centuries of Citations
Cannabis has been utilized in one form or another for treatment of pain for longer than written history [18–21]. Although this documentation has been a major preoccupation of the lead author [22–25], and such information can provide provocative direction to inform modern research on treatment of pain and other conditions, it does not represent evidence of form, content, or degree that is commonly acceptable to governmental regulatory bodies with respect to pharmaceutical development.
Anecdotes Versus Modern Proof of Concept
While thousands of compelling stories of efficacy of cannabis in pain treatment certainly underline the importance of properly harnessing cannabinoid mechanisms therapeutically [26, 27], prescription analgesics in the United States necessitate Food and Drug Administration (FDA) approval. This requires a rigorous development program proving consistency, quality, efficacy, and safety as defined by basic scientific studies and randomized controlled trials (RCT) [28] and generally adhering to recent IMMPACT recommendations [29], provoking our next question.
Can a Botanical Agent Become a Prescription Medicine?
Most modern physicians fail to recognize that pharmacognosy (study of medicinal plants) has led directly or indirectly to an estimated 25 % of modern pharmaceuticals [30]. While the plethora of available herbal agents yield an indecipherable cacophony to most clinicians and consumers alike, it is certainly possible to standardize botanical agents and facilitate their recommendation based on sound science [31]. Botanical medicines can even fulfill the rigorous dictates of the FDA and attain prescription drug status via a clear roadmap in the form of a blueprint document [32], henceforth termed the Botanical Guidance: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070491.pdf. To be successful and clinically valuable, botanicals, including cannabis-based medicines, must demonstrate the same quality, clinical analgesic benefit, and appropriately safe adverse event profile as available new chemical entities (NCE) [28].
The Biochemical and Neurophysiological Basis of Pain Control by Cannabinoids
Neuropathic Pain
Thorough reviews of therapeutic effects of cannabinoids in preclinical and clinical domains have recently been published [33, 34]. In essence, the endocannabinoid system (ECS) is active throughout the CNS and PNS in modulating pain at spinal, supraspinal, and peripheral levels. Endocannabinoids are produced on demand in the CNS to dampen sensitivity to pain [35]. The endocannabinoid system is operative in such key integrative pain centers as the periaqueductal grey matter [36, 37], the ventroposterolateral nucleus of the thalamus [38], and the spinal cord [39, 40]. Endocannabinoids are endogenous mediators of stress-induced analgesia and fear-conditioned analgesia and suppress pain-related phenomena such as windup [41] and allodynia [42]. In the periphery and PNS [13], the ECS has key effects in suppressing both hyperalgesia and allodynia via CB1 [43] and CB2 mechanisms (Fig. 18.2). Indeed, pathological pain states have been postulated to arise, at least in part, from a dysregulation of the endocannabinoid system.
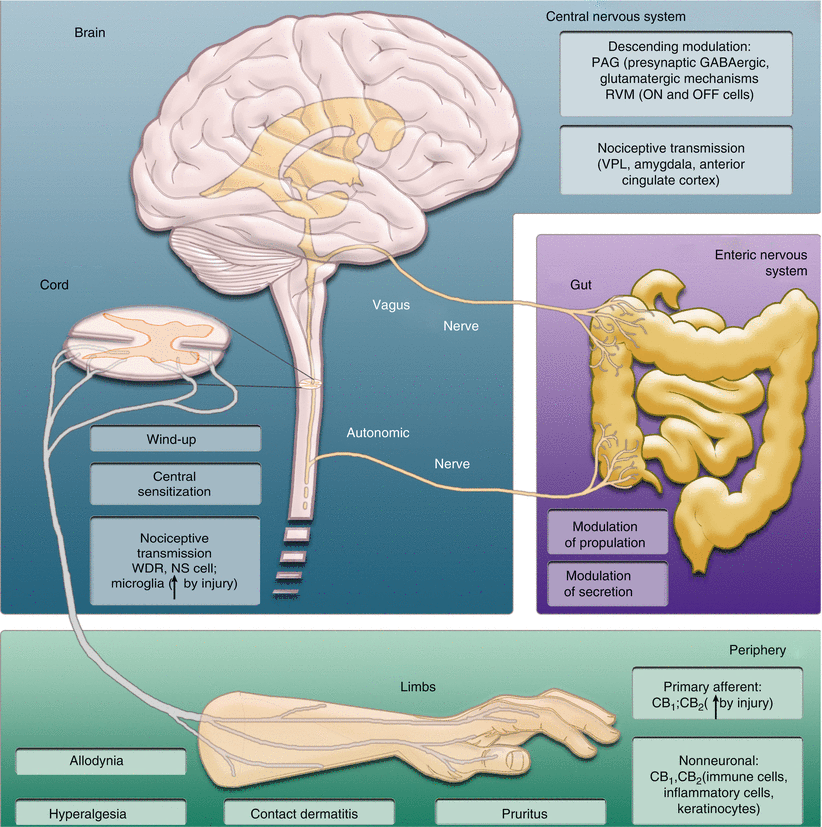
Fig. 18.2
Cannabinoids suppress pain and other pathophysiological (e.g., contact dermatitis, pruritis) and physiological (e.g., gastrointestinal transit and secretion) processes through multiple mechanisms involving CB1 and CB2 receptors. Peripheral, spinal, and supraspinal sites of cannabinoid actions are shown. In the periphery, cannabinoids act through both neuronal and nonneuronal mechanisms to control inflammation, allodynia, and hyperalgesia. CB1 and CB2 have been localized to both primary afferents and nonneuronal cells (e.g., keratinocytes, microglia), and expression can be regulated by injury. In the spinal cord, cannabinoids suppress nociceptive transmission, windup, and central sensitization by modulating activity in the ascending pain pathway of the spinothalamic tract, including responses of wide dynamic range (WDR) and nociceptive specific (NS) cells. Similar processes are observed at rostral levels of the neuraxis (e.g., ventroposterolateral nucleus of the thalamus, amygdala, anterior cingulate cortex). Cannabinoids also actively modulate pain through descending mechanisms. In the periaqueductal gray, cannabinoids act through presynaptic glutamatergic and GABAergic mechanisms to control nociception. In the rostral ventromedial medulla, cannabinoids suppress activity in ON cells and inhibit the firing pause of OFF cells, in response to noxious stimulation to produce antinociception (Figure by authors with kind assistance of James Brodie, GW Pharmaceuticals)
Antinociceptive and Anti-inflammatory Pain Mechanisms
Beyond the mechanisms previously mentioned, the ECS plays a critical role in peripheral pain, inflammation, and hyperalgesia [43] through both CB1 and CB2 mechanisms. CB1 and CB2 mechanisms are also implicated in regulation of contact dermatitis and pruritus [44]. A role for spinal CB2 mechanisms, mediated by microglia and/or astrocytes, is also revealed under conditions of inflammation [45]. Both THC and cannabidiol (CBD), a non-euphoriant phytocannabinoid common in certain cannabis strains, are potent anti-inflammatory antioxidants with activity exceeding that of vitamins C and E via non-cannabinoid mechanisms [46]. THC inhibits prostaglandin E-2 synthesis [47] and stimulates lipooxygenase [48]. Neither THC nor CBD affects COX-1 or COX-2 at relevant pharmacological dosages [49].
While THC is inactive at vanilloid receptors, CBD, like AEA, is a TRPV1 agonist. Like capsaicin, CBD is capable of inhibiting fatty-acid amide hydrolase (FAAH), the enzyme which hydrolyzes AEA and other fatty-acid amides that do not bind to cannabinoid receptors. CBD additionally inhibits AEA reuptake [50] though not potently. Thus, CBD acts as an endocannabinoid modulator [51], a mechanism that various pharmaceutical firms hope to emulate with new chemical entities (NCEs). CBD inhibits hepatic metabolism of THC to 11-hydroxy-THC, which is possibly more psychoactive, and prolongs its half-life, reducing its psychoactivity and attenuating attendant anxiety and tachycardia [51]; antagonizes psychotic symptoms [52]; and attenuates appetitive effects of THC [53] as well as its effects on short-term memory [54]. CBD also inhibits tumor necrosis factor-alpha (TNF-α) in a rodent model of rheumatoid arthritis [55]. Recently, CBD has been demonstrated to enhance adenosine receptor A2A signaling via inhibition of the adenosine transporter [56].
Recently, GPR18 has been proposed as a putative CBD receptor whose function relates to cellular migration [57]. Antagonism of GPR18 (by agents such as CBD) may be efficacious in treating pain of endometriosis, among other conditions, especially considering that such pain may be endocannabinoid-mediated [58]. Cannabinoids are also very active in various gastrointestinal and visceral sites mediating pain responses [59, 60].
Cannabinoid Interactions with Other Neurotransmitters Pertinent to Pain
As alluded to above, the ECS modulates neurotransmitter release via retrograde inhibition. This is particularly important in NMDA-glutamatergic mechanisms that become hyperresponsive in chronic pain states. Cannabinoids specifically inhibit glutamate release in the hippocampus [61]. THC reduces NMDA responses by 30–40 % [46]. Secondary and tertiary hyperalgesia mediated by NMDA [62] and by calcitonin gene-related peptide [40] may well be targets of cannabinoid therapy in disorders such as migraine, fibromyalgia, and idiopathic bowel syndrome wherein these mechanisms seem to operate pathophysiologically [63], prompting the hypothesis of a “clinical endocannabinoid deficiency.” Endocannabinoid modulators may therefore restore homeostasis, leading to normalization of function in these pathophysiological conditions. THC also has numerous effects on serotonergic systems germane to migraine [64], increasing its production in the cerebrum while decreasing reuptake [65]. In fact, the ECS seems to modulate the trigeminovascular system of migraine pathogenesis at vascular and neurochemical levels [66–68].
Cannabinoid-Opioid Interactions
Although endocannabinoids do not bind to opioid receptors, the ECS may nonetheless work in parallel with the endogenous opioid system with numerous areas of overlap and interaction. Pertinent mechanisms include stimulation of beta-endorphin by THC [69] as well as its ability to demonstrate experimental opiate sparing [70], prevent opioid tolerance and withdrawal [71], and rekindle opioid analgesia after loss of effect [72]. Adjunctive treatments that combine opioids with cannabinoids may enhance the analgesic effects of either agent. Such strategies may permit lower doses of analgesics to be employed for therapeutic benefit in a manner that minimizes incidence or severity of adverse side effects.
Clinical Trials, Utility, and Pitfalls of Cannabinoids in Pain
Evidence for Synthetic Cannabinoids
Oral dronabinol (THC) has been available as the synthetic Marinol® since 1985 and is indicated for nausea associated with chemotherapy and appetite stimulation in HIV/AIDS. Issues with its cost, titration difficulties, delayed onset, and propensity to induce intoxicating and dysphoric effects have limited clinical application [73]. It was employed in two open-label studies of chronic neuropathic pain in case studies in 7 [74] and 8 patients [75], but no significant benefit was evident and side effects led to prominent dropout rates (average doses 15–16.6 mg THC). Dronabinol produced benefit in pain in multiple sclerosis [76], but none was evident in postoperative pain (Table 18.1) [77]. Dronabinol was reported to relieve pruritus in three case-report subjects with cholestatic jaundice [78]. Dronabinol was assessed in 30 chronic non-cancer pain patients on opioids in double-blind crossover single-day sessions vs. placebo with improvement [79], followed by a 4-week open-label trial with continued improvement (Table 18.1). Associated adverse events were prominent. Methodological issues included lack of prescreening for cannabinoids, 4 placebo subjects with positive THC assays, and 58 % of subjects correctly guessing Marinol dose on test day. An open-label comparison in polyneuropathy examined nabilone patients with 6 obtaining 22.6 % mean pain relief after 3 months, and 5 achieving 28.6 % relief after 6 months, comparable to conventional agents [80]. A pilot study of Marinol in seven spinal cord injury patients with neuropathic pain saw two withdraw, and the remainder appreciate no greater efficacy than with diphenhydramine [81].
Table 18.1
Randomized controlled trials of cannabinoids in pain
Agent | N = | Indication | Duration/type | Outcomes/reference |
|---|---|---|---|---|
Ajulemic acid | 21 | Neuropathic pain | 7 day crossover | Visual analogue pain scales improved over placebo (p = 0.02)/Karst et al. [92] |
Cannabis, smoked | 50 | HIV neuropathy | 5 days/DB | Decreased daily pain (p = 0.03) and hyperalgesia (p = 0.05), 52 % with >30 % pain reduction vs. placebo (p = 0.04)/Abrams et al. [94] |
Cannabis, smoked | 23 | Chronic neuropathic pain | 5 days/DB | Decreased pain vs. placebo only at 9.4 % THC level (p = 0.023)/Ware et al. [98] |
Cannabis, smoked | 38 | Neuropathic pain | Single dose/DBC | NSD in pain except at highest cannabis dose (p = 0.02), with prominent psychoactive effects/Wilsey et al. [95] |
Cannabis, smoked | 34 | HIV neuropathy | 5 days /DB | DDS improved over placebo (p = 0.016), 46 % vs. 18 % improved >30 %, 2 cases toxic psychosis/Ellis et al. [97] |
Cannabis, vaporized | 21 | Chronic pain on opioids | 5 days/DB | 27 % decrement in pain/Abrams et al. [118] |
Cannador | 419 | Pain due to spasm in MS | 15 weeks | Improvement over placebo in subjective pain associated with spasm (p = 0.003)/Zajicek et al. [120] |
Cannador | 65 | Postherpetic neuralgia | 4 weeks | No benefit observed/Ernst et al. [122] |
Cannador | 30 | Postoperative pain | Single doses, daily | Decreasing pain intensity with increased dose (p = 0.01)/Holdcroft et al. [123] |
Marinol | 24 | Neuropathic pain in MS | 15–21 days/DBC | Median numerical pain (p = 0.02), median pain relief improved (p = 0.035) over placebo/Svendsen et al. [76] |
Marinol | 40 | Postoperative pain | Single dose/DB | No benefit observed over placebo/Buggy et al. [77] |
Marinol | 30 | Chronic pain | 3 doses, 1 day/DBC | Total pain relief improved with 10 mg (p < 0.05) and 20 mg (p < 0.01) with opioids, AE prominent/Narang et al. [79] |
Nabilone | 41 | Postoperative pain | 3 doses in 24 h/DB | NSD morphine consumption. Increased pain at rest and on movement with nabilone 1 or 2 mg/Beaulieu [85] |
Nabilone | 31 | Fibromyalgia | 2 weeks/DBC | Compared to amitriptyline, nabilone improved sleep, decrease wakefulness, had no effect on pain, and increased AE/Ware et al. [90] |
Nabilone | 96 | Neuropathic pain | 14 weeks/DBC vs. dihydrocodeine | Dihydrocodeine more effective with fewer AE/Frank et al. [88] |
Nabilone | 13 | Spasticity pain | 9 weeks/DBC | NRS decreased 2 points for nabilone (p < 0.05)/Wissel et al. [87] |
Nabilone | 40 | Fibromyalgia | 4 weeks/DBC | VAS decreased in pain, Fibromyalgia Impact Questionnaire, and anxiety over placebo (all, p < 0.02)/Skrabek et al. [89] |
Sativex | 20 | Neurogenic pain | Series of 2-week N-of-1 crossover blocks | Improvement with Tetranabinex and Sativex on VAS pain vs. placebo (p < 0.05), symptom control best with Sativex (p < 0.0001)/Wade et al. [132] |
Sativex | 24 | Chronic intractable pain | 12 weeks, series of N-of-1 crossover blocks | |
Sativex | 48 | Brachial plexus avulsion | 6 weeks in 3 two-week crossover blocks | Benefits noted in Box Scale-11 pain scores with Tetranabinex (p = 0.002) and Sativex (p = 0.005) over placebo/Berman et al. [134] |
Sativex | 66 | Central neuropathic pain in MS | 5 weeks | Numerical Rating Scale (NRS) analgesia improved over placebo (p = 0.009)/Rog et al. [135] |
Sativex | 125 | Peripheral neuropathic pain | 5 weeks | Improvements in NRS pain levels (p = 0.004), dynamic allodynia (p = 0.042), and punctuate allodynia (p = 0.021) vs. placebo/Nurmikko et al. [136] |
Sativex | 56 | Rheumatoid arthritis | Nocturnal dosing for 5 weeks | Improvements over placebo morning pain on movement (p = 0.044), morning pain at rest (p = 0.018), DAS-28 (p = 0.002), and SF-MPQ pain at present (p = 0.016)/Blake et al. [138] |
Sativex | 117 | Pain after spinal injury | 10 days | NSD in NRS pain scores, but improved Brief Pain Inventory (p = 0.032), and Patients’ Global Impression of Change (p = 0.001) (unpublished) |
Sativex | 177 | Intractable cancer pain | 2 weeks | Improvements in NRS analgesia vs. placebo (p = 0.0142), Tetranabinex NSD/Johnson et al. [139] |
Sativex | 135 | Intractable lower urinary tract symptoms in MS | 8 weeks | Improved bladder severity symptoms including pain over placebo (p = 0.001) [200] |
Sativex | 360 | Intractable cancer pain | 5 weeks/DB | CRA of lower and middle-dose cohorts improved over placebo (p = 0.006)/ [201] |
Nabilone, or Cesamet®, is a semisynthetic analogue of THC that is about tenfold more potent, and longer lasting [82]. It is indicated as an antiemetic in chemotherapy in the USA. Prior case reports in neuropathic pain [83] and other pain disorders [84] have been published. Sedation and dysphoria are prominent associated adverse events. An RCT of nabilone in 41 postoperative subjects dosed TID actually resulted in increased pain scores (Table 18.1) [85]. An uncontrolled study of 82 cancer patients on nabilone noted improved pain scores [86], but retention rates were limited. Nabilone improved pain (p < 0.05) vs. placebo in patients with mixed spasticity syndromes in a small double-blind trial (Table 18.1) [87], but was without benefits in other parameters. In a double-blind crossover comparison of nabilone to dihydrocodeine (schedule II opioid) in chronic neuropathic pain (Table 18.1) [88], both drugs produced marginal benefit, but with dihydrocodeine proving clearly superior in efficacy and modestly superior in side-effect profile. In an RCT in 40 patients of nabilone vs. placebo over 4 weeks, it showed significant decreases in VAS of pain and anxiety (Table 18.1) [89]. A more recent study of nabilone vs. amitriptyline in fibromyalgia yielded benefits on sleep, but not pain, mood, or quality of life (Table 18.1) [90]. An open-label trial of nabilone vs. gabapentin found them comparable in pain and other symptom relief in peripheral neuropathic pain [91].
Evidence for Smoked or Vaporized Cannabis
Few randomized controlled clinical trials (RCTs) of pain with smoked cannabis have been undertaken to date [94–97]. One of these [96] examined cannabis effects on experimental pain in normal volunteers.
Abrams et al. [94] studied inpatient adults with painful HIV neuropathy in 25 subjects in double-blind fashion to receive either smoked cannabis as 3.56 % THC cigarettes or placebo cigarettes three times daily for 5 days (Table 18.1). The smoked cannabis group had a 34 % reduction in daily pain vs. 17 % in the placebo group (p = 0.03). The cannabis cohort also had a 52 % of subjects report a >30 % reduction in pain scores over the 5 days vs. 24 % in the placebo group (p = 0.04) (Table 18.1). The authors rated cannabis as “well tolerated” due to an absence of serious adverse events (AE) leading to withdrawal, but all subjects were cannabis experienced. Symptoms of possible intoxication in the cannabis group including anxiety (25 %), sedation (54 %), disorientation (16 %), paranoia (13 %), confusion (17 %), dizziness (15 %), and nausea (11 %) were all statistically significantly more common than in the placebo group. Despite these findings, the authors stated that the values do not represent any serious safety concern in this short-term study. No discussion in the article addressed issues of the relative efficacy of blinding in the trial.
Wilsey et al. [95] examined neuropathic pain in 38 subjects in a double-blind crossover study comparing 7 % THC cannabis, 3.5 % THC cannabis, and placebo cigarettes via a complex cumulative dosing scheme with each dosage given once, in random order, with at least 3 day intervals separating sessions (Table 18.1). A total of 9 puffs maximum were allowed over several hours per session. Authors stated, “Psychoactive effects were minimal and well-tolerated, but neuropsychological impairment was problematic, particularly with the higher concentration of study medication.” Again, only cannabis-experienced subjects were allowed entry. No withdrawals due to AE were reported, but 1 subject was removed due to elevated blood pressure. No significant differences were noted in pain relief in the two cannabis potency groups, but a significant separation of pain reduction from placebo (p = 0.02) was not evident until a cumulative 9 puffs at 240 min elapsed time. Pain unpleasantness was also reduced in both active treatment groups (p < 0.01). Subjectively, an “any drug effect” demonstrated a visual analogue scale (VAS) of 60/100 in the high-dose group, but even the low-dose group registered more of a “good drug effect” than placebo (p < 0.001). “Bad drug effect” was also evident. “Feeling high” and “feeling stoned” were greatest in the high-dose sessions (p < 0.001), while both high- and low-dose differentiated significantly from placebo (p < 0.05). Of greater concern, both groups rated impairment as 30/100 on VAS vs. placebo (p = 0.003). Sedation also demarcated both groups from placebo (p < 0.01), as did confusion (p = 0.03), and hunger (p < 0.001). Anxiety was not considered a prominent feature in this cannabis-experienced population. This study distinguished itself from some others in its inclusion of specific objective neuropsychological measures and demonstrated neurocognitive impairment in attention, learning, and memory, most noteworthy with 7 % THC cannabis. No commentary on blinding efficacy was included.
Ellis et al. [97] examined HIV-associated neuropathic pain in a double-blind trial of placebo vs. 1–8 % THC cannabis administered four times daily over 5 days with a 2-week washout (Table 18.1). Subjects were started at 4 % THC and then titrated upward or downward in four smoking sessions dependent upon their symptom relief and tolerance of the dose. In this study, 96 % of subjects were cannabis-experienced, and 28 out of 34 subjects completed the trial. The primary outcome measure (Descriptor Differential Scale, DDS) was improved in the active group over placebo (p = 0.016), with >30 % relief noted in 46 % of cannabis subjects vs. 18 % of placebo. While most adverse events (AE) were considered mild and self-limited, two subjects had to leave the trial due to toxicity. One cannabis-naïve subject was withdrawn due to “an acute cannabis-induced psychosis” at what proved to be his first actual cannabis exposure. The other subject suffered intractable cough. Pain reduction was greater in the cannabis-treated group (p = 0.016) among completers, as was the proportion of subjects attaining >30 % pain reduction (46 % vs. 18 %, p = 0.043). Blinding was assessed in this study; whereas placebo patients were inaccurate at guessing the investigational product, 93 % of those receiving cannabis guessed correctly. On safety issues, the authors stated that the frequency of some nontreatment-limiting side effects was greater for cannabis than placebo. These included concentration difficulties, fatigue, sleepiness or sedation, increased duration of sleep, reduced salivation, and thirst.
A Canadian study [98] examined single 25-mg inhalations of various cannabis potencies (0–9.4 % THC) three times daily for 5 days per cycle in 23 subjects with chronic neuropathic pain (Table 18.1). Patients were said to be cannabis-free for 1 year, but were required to have some experience of the drug. Only the highest potency demarcated from placebo on decrements in average daily pain score (5.4 vs. 6.1, p = 0.023). The most frequent AE in the high-dose group were headache, dry eyes, burning sensation, dizziness, numbness, and cough, but with “high” or “euphoria” reported only once in each cannabis potency group.
The current studies of smoked cannabis are noteworthy for their extremely short-term exposure and would be of uncertain relevance in a regulatory environment. The IMMPACT recommendations on chronic neuropathic pain clinical trials that are currently favored by the FDA [29] generally suggest randomized controlled clinical trials of 12-week duration as a prerequisite to demonstrate efficacy and safety. While one might assume that the degree of pain improvement demonstrated in these trials could be maintained over this longer interval, it is only reasonable to assume that cumulative adverse events would also increase to at least some degree. The combined studies represent only a total of 1,106 patient-days of cannabis exposure (Abrams: 125, Wilsey: 76, Ellis: 560, Ware 345) or 3 patient-years of experience. In contrast, over 6,000 patient-years of data have been analyzed for Sativex between clinical trials, prescription, and named-patient supplies, with vastly lower AE rates (data on file, GW Pharmaceuticals) [28, 99]. Certainly, the cognitive effects noted in California-smoked cannabis studies figure among many factors that would call the efficacy of blinding into question for investigations employing such an approach. However, it is also important to emphasize that unwanted side effects are not unique to cannabinoids. In a prospective evaluation of specific chronic polyneuropathy syndromes and their response to pharmacological therapies, the presence of intolerable side effects did not differ in groups receiving gabapentinoids, tricyclic antidepressants, anticonvulsants, cannabinoids (including nabilone, Sativex), and topical agents [80]. Moreover, no serious adverse events were related to any of the medications.
The current studies were performed in a very select subset of patients who almost invariably have had prior experience of cannabis. Their applicability to cannabis-naïve populations is, thus, quite unclear. At best, the observed benefits might possibly accrue to some, but it is eminently likely that candidates for such therapy might refuse it on any number of grounds: not wishing to smoke, concern with respect to intoxication, etc. Sequelae of smoking in therapeutic outcomes have had little discussion in these brief RCTs [28]. Cannabis smoking poses substantial risk of chronic cough and bronchitic symptoms [100], if not obvious emphysematous degeneration [101] or increase in aerodigestive cancers [102]. Even such smoked cannabis proponents as Lester Grinspoon has acknowledged are the only well-confirmed deleterious physical effect of marihuana is harm to the pulmonary system [103]. However, population-based studies of cannabis trials have failed to show any evidence for increased risk of respiratory symptoms/chronic obstructive pulmonary disease [100] or lung cancer [102] associated with smoking cannabis.
A very detailed analysis and comparison of mainstream and sidestream smoke for cannabis vs. tobacco smoke was performed in Canada [104]. Of note, cannabis smoke contained ammonia (NH3) at a level of 720 μg per 775 mg cigarette, a figure 20-fold higher than that found in tobacco smoke. It was hypothesized that this finding was likely attributable to nitrate fertilizers. Formaldehyde and acetaldehyde were generally lower in cannabis smoke than in tobacco, but butyraldehyde was higher. Polycyclic aromatic hydrocarbon (PAH) contents were qualitatively similar in the comparisons, but total yield was lower for cannabis mainstream smoke, but higher than tobacco for sidestream smoke. Additionally, NO, NO x , hydrogen cyanide, and aromatic amines concentrations were 3–5 times higher in cannabis smoke than that from tobacco. Possible mutagenic and carcinogenic potential of these various compounds were mentioned. More recently, experimental analysis of cannabis smoke with resultant acetaldehyde production has posited its genotoxic potential to be attributable to reactions that produce DNA adducts [105].
Vaporizers for cannabis have been offered as a harm reduction technique that would theoretically eliminate products of combustion and associated adverse events. The Institute of Medicine (IOM) examined cannabis issues in 1999 [106], and among their conclusions was the following (p. 4): “Recommendation 2: Clinical trials of cannabinoid drugs for symptom management should be conducted with the goal of developing rapid-onset, reliable, and safe delivery systems.” One proposed technique is vaporization, whereby cannabis is heated to a temperature that volatilizes THC and other components with the goal of reducing or eliminating by-products of combustion, including potentially carcinogenic polycyclic aromatic hydrocarbons, benzene, acetaldehyde, carbon monoxide, toluene, naphthaline, phenol, toluene, hydrogen cyanide, and ammonia. Space limitations permit only a cursory review of available literature [107–115].
A pilot study of the Volcano vaporizer vs. smoking was performed in the USA in 2007 in 18 active cannabis consumers, with only 48 h of presumed abstinence [116]. NIDA 900-mg cannabis cigarettes were employed (1.7, 3.4, and 6.8 % THC) with each divided in two, so that one-half would be smoked or vaporized in a series of double-blind sessions. The Volcano vaporizer produced comparable or slightly higher THC plasma concentrations than smoking. Measured CO in exhaled vapor sessions diminished very slightly, while it increased after smoking (p < 0.001). Self-reported visual analogue scales of the associated high were virtually identical in vaporization vs. smoking sessions and increased with higher potency material. A contention was advanced that the absence of CO increase after vaporization can be equated to “little or no exposure to gaseous combustion toxins.” Given that no measures of PAH or other components were undertaken, the assertion is questionable. It was also stated that there were no reported adverse events. Some 12 subjects preferred the Volcano, 2 chose smoking, and 2 had no preference as to technique, making the vaporizer “an acceptable system” and providing “a safer way to deliver THC.”
A recent [202, 117] examined interactions of 3.2 % THC NIDA cannabis vaporized in the Volcano in conjunction with opioid treatment in a 5-day inpatient trial in 21 patients with chronic pain (Table 18.1). All subjects were prior cannabis smokers. Overall, pain scores were reduced from 39.6 to 29.1 on a VAS, a 27 % reduction, by day 5. Pain scores in subjects on morphine fell from 34.8 to 24.1, while in subjects taking oxycodone, scores dropped from 43.8 to 33.6.
The clinical studies performed with vaporizers to date have been very small pilot studies conducted over very limited timeframes (i.e., for a maximum of 5 days). Thus, these studies cannot contribute in any meaningful fashion toward possible FDA approval of vaporized cannabis as a delivery technique, device, or drug under existing policies dictated by the Botanical Guidance [32]. It is likewise quite unlikely that the current AE profile of smoked or vaporized cannabis would meet FDA requirements. The fact that all the vaporization trials to date have been undertaken only in cannabis-experienced subjects does not imply that results would generalize to larger patient populations. Moreover, there is certainly no reason to expect AE profiles to be better in cannabis-naïve patients. Additionally, existing standardization of cannabis product and delivery via vaporization seem far off the required marks. Although vaporizers represent an alternate delivery method devoid of the illegality associated with smoked cannabis, the presence of toxic ingredients such as PAH, ammonia, and acetaldehyde in cannabis vapor are unlikely to be acceptable to FDA in any significant amounts. Existing vaporizers still lack portability or convenience [28]. A large Internet survey revealed that only 2.2 % of cannabis users employed vaporization as their primary cannabis intake method [118]. While studies to date have established that lower temperature vaporization in the Volcano, but not necessarily other devices, can reduce the relative amounts of noxious by-products of combustion, it has yet to be demonstrated that they are totally eliminated. Until or unless this goal is achieved, along with requisite benchmarks of herbal cannabis quality, safety, and efficacy in properly designed randomized clinical trials, vaporization remains an unproven technology for therapeutic cannabinoid administration.
Evidence for Cannabis-Based Medicines
Cannador is a cannabis extract in oral capsules, with differing THC:CBD ratios [51]. Cannador was utilized in a phase III RCT of spasticity in multiple sclerosis (CAMS) (Table 18.1) [119]. While no improvement was evident in the Ashworth Scale, reduction was seen in spasm-associated pain. Both THC and Cannador improved pain scores in follow-up [120]. Cannador was also employed for postherpetic neuralgia in 65 patients, but without success (Table 18.1) [121, 122]. Slight pain reduction was observed in 30 subjects with postoperative pain (CANPOP) not receiving opiates, but psychoactive side effects were notable (Table 18.1).
Sativex® is a whole-cannabis-based extract delivered as an oromucosal spray that combines a CB1 and CB2 partial agonist (THC) with a cannabinoid system modulator (CBD), minor cannabinoids, and terpenoids plus ethanol and propylene glycol excipients and peppermint flavoring [51, 123]. It is approved in Canada for spasticity in MS and under a Notice of Compliance with Conditions for central neuropathic pain in multiple sclerosis and treatment of cancer pain unresponsive to opioids. Sativex is also approved in MS in the UK, Spain, and New Zealand, for spasticity in multiple sclerosis, with further approvals expected soon in some 22 countries around the world. Sativex is highly standardized and is formulated from two Cannabis sativa chemovars predominating in THC and CBD, respectively [124]. Each 100 μl pump-action oromucosal spray of Sativex yields 2.7 mg of THC and 2.5 mg of CBD plus additional components. Pharmacokinetic data are available [125–127]. Sativex effects begin within an interval allowing dose titration. A very favorable adverse event profile has been observed in the development program [27, 128]. Most patients stabilize at 8–10 sprays per day after 7–10 days, attaining symptomatic control without undue psychoactive sequelae. Sativex was added to optimized drug regimens in subjects with uncontrolled pain in every RCT (Table 18.1). An Investigational New Drug (IND) application to study Sativex in advanced clinical trials in the USA was approved by the FDA in January 2006 in patients with intractable cancer pain. One phase IIB dose-ranging study has already been completed [201]. Available clinical trials with Sativex have been independently assessed [129, 130].
In a phase II study of 20 patients with neurogenic symptoms [131], significant improvement was seen with both Tetranabinex (high-THC extract without CBD) and Sativex on pain, with Sativex displaying better symptom control (p < 0.0001), with less intoxication (Table 18.1).
In a phase II study of intractable chronic pain in 24 patients [132], Sativex again produced the best results compared to Tetranabinex (p < 0.001), especially in MS (p < 0.0042) (Table 18.1).
In a phase III study of brachial plexus avulsion (N = 48) [133], pain reduction with Tetranabinex and Sativex was about equal (Table 18.1).
In an RCT of 66 MS subjects, mean Numerical Rating Scale (NRS) analgesia favored Sativex over placebo (Table 18.1) [134].
In a phase III trial (N = 125) of peripheral neuropathic pain with allodynia [135], Sativex notably alleviated pain levels and dynamic and punctate allodynia (Table 18.1).
In a safety-extension study in 160 subjects with various symptoms of MS [136], 137 patients showed sustained improvements over a year or more in pain and other symptoms [99] without development of any tolerance requiring dose escalation or withdrawal effects in those who voluntarily discontinued treatment suddenly. Analgesia was quickly reestablished upon Sativex resumption.
In a phase II RCT in 56 rheumatoid arthritis sufferers over 5 weeks with Sativex [137], medicine was limited to only 6 evening sprays (16.2 mg THC + 15 mg CBD). By study end, morning pain on movement, morning pain at rest, DAS-28 measure of disease activity, and SF-MPQ pain all favored Sativex (Table 18.1).
In a phase III RCT in intractable cancer pain on opioids (N = 177), Sativex, Tetranabinex THC-predominant extract, and placebo were compared [138] demonstrating strongly statistically significant improvements in analgesia for Sativex only (Table 18.1). This suggests that the CBD component in Sativex was necessary for benefit.
In a 2-week study of spinal cord injury pain, NRS of pain was not statistically different from placebo, probably due to the short duration of the trial, but secondary endpoints were positive (Table 18.1). Additionally, an RCT of intractable lower urinary tract symptoms in MS also demonstrated pain reduction (Table 18.1).
The open-label study of various polyneuropathy patients included Sativex patients with 3 obtaining 21.56 % mean pain relief after 3 months (2/3 > 30 %), and 4 achieving 27.6 % relief after 6 months (2/4 > 30 %), comparable to conventional agents [80].
A recently completed RCT of Sativex in intractable cancer pain unresponsive to opioids over 5 weeks was performed in 360 subjects (Table 18.1). Results of a Continuous Response Analysis (CRA) showed improvements over placebo in the low-dose (p = 0.08) and middle-dose cohorts (p = 0.038) or combined (p = 0.006). Pain NRS improved over placebo in the low-dose (p = 0.006) and combined cohorts (p = 0.019).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


