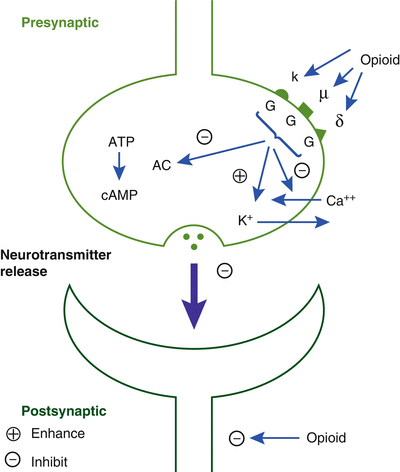
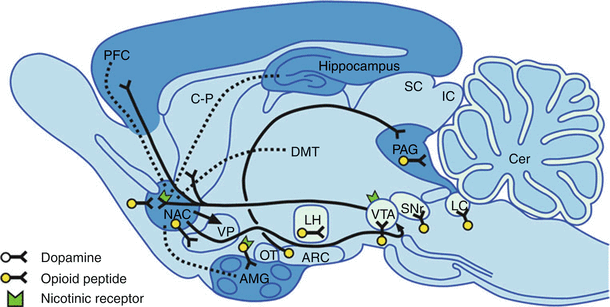
The dopaminergic system in the ventral tegmental area (VTA) is the site of the natural reward centers of the brain, and GABA neurons usually inhibit these dopaminergic systems. Opioids inhibit the presynaptic receptors on these GABA neurons, which increases the release of dopamine, which is intensely pleasurable. These are the same areas of the brain associated with other drugs of abuse such as alcohol, nicotine, and benzodiazepines (Fig. 10.3) [10].
Opioid Genetics
Each opioid receptor has a different activity, as well as a different receptor affinity (which is genetically controlled). For example, OPRM1, the gene that encodes the mu receptor, is polymorphic, and approximately 20–30 % of the population has heterozygous changes in the alleles, associated with altered sensitivities to pain [11]. Different opioids also have different relative affinity for each receptor, so that the same opioid may have very different effects on different people, and the same person might have different effects from different opioids.
There is now considerable evidence suggesting genetic variability in the ability of individuals to metabolize and respond to drugs. All opioid drugs are substantially metabolized, mainly by the cytochrome P450 system as well as to a lesser degree the UDP-glucuronosyltransferase system (UGTs). Activity of these enzymes depends on whether patient is homozygous for nonfunctioning alleles (poor metabolizer or PM), has at least one functioning allele (extensive metabolizer or EM), or has multiple copies of a functional allele (ultrarapid metabolizer UM) [12].
As a result, the morphine dose needed for postoperative pain relief after similar surgery may vary fivefold between individuals, and the dose needed at a defined stage of cancer pain varies threefold [13]. As another example, CYP 450 2D6 is a critical enzyme involved in the metabolism of a variety of opioids described below; activity of this enzyme is highly variable, and there may be as much as a 10,000-fold difference among individuals [14]. Approximately 8–10 % of Caucasians but up to 50 % of people of Asian descent have an inactive form of this enzyme [15]. As discussed below, hydrocodone is metabolized to hydromorphone via CYP 2D6; in one study [16], the metabolism of hydrocodone to hydromorphone was eight times faster in EMs than in PMs. Medications may also interfere with enzyme activity; in this same study, quinidine, a potent CYP2D6 inhibitor, reduced the excretion of hydromorphone, resulting in plasma levels five times higher in EMs than PMs.
Opioid Side Effects
Opioids are well known to cause a variety of side effects, most commonly nausea and vomiting, constipation, sedation, and respiratory depression [17]. These side effects can be significant, and some patients avoid opioids even in the face of significant pain, in an effect to limit such side effects, which may act as a significant barrier to adequate pain relief [18].
Constipation
Constipation is the most common adverse effect from opioids, occurring in 40–95 % of patients treated with opioids [19], and is caused by opioid receptor stimulation in the gut. The subsequent decrease in GI motility results in increased fecal fluid absorption, resulting in hard, dry stools. It is essential that prophylactic treatment be instituted on the initiation of opioid treatment since this, of all the side effects of opioids, does not resolve over time.
Nausea
Pruritus
Two to ten percent of patients on opioids will develop pruritus [18], which results from a direct release of histamine and not usually an antigen/antibody reaction. It is therefore better considered an adverse reaction than an allergic reaction and is usually treated symptomatically with antihistamines such as diphenhydramine.
Sedation and Cognitive Dysfunction
Respiratory Depression
A significant proportion of patients taking long-term opioids develop central apnea during sleep. Teichtahl and colleagues [24] examined ten patients in a methadone maintenance program and performed a clinical assessment and overnight polysomnography. They found that all ten patients had evidence of central sleep apnea, with six patients having a central apnea index (CAI) [the number of central apnea events per hour] [25] greater than 5, and four patients with a CAI greater than 10. In a larger follow-up study of 50 patients taking long-term methadone, 30 % of the patients had a CAI greater than 5, and 20 % had a CAI greater than 10 [26].
Endocrine Effects
Endorphins appear to be primarily involved in the regulation of gonadotropins and ACTH release [27]. Amenorrhea developed in 52 % female patients on opioids for chronic pain [28], while the testosterone levels were subnormal in 74 % of males on sustained-release oral opioids [29]. These effects are more profound with IV or intrathecal opioids than oral opioids [30].
Immunologic Effects
Acute and chronic opioid administration can cause inhibitory effects on antibody and cellular immune responses, natural killer cell activity, cytokine expression, and phagocytic activity. Chronic administration of opioids decreases the proliferative capacity of macrophage progenitor cells and lymphocytes [31].
Relationship Between Side Effects and Sex or Ethnicity
Several studies suggest that sex and ethnic differences exist to explain the differences seen in side effect profiles. Women have, for instance, been found to be more sensitive to the respiratory effects of morphine [32] and more often have nausea and emesis with opioids [33]. Varying levels of opioid metabolites due to genetic differences in CYP 450 isoenzymes and glucuronidation between ethnic groups [34] may explain the variety of responses seen to similar doses of medications (see section “Opioid Metabolism”).
Opioid Metabolism
Many of the positive effects, as well as the side effects of opioids, can be traced to their metabolites, and knowledge of these metabolites may help to explain many of the puzzling clinical scenarios seen by the practicing physician.
Morphine
Morphine is metabolized by glucuronidation, producing morphine-6-glucuronide (M6G) and morphine-3-glucuronide (M3G) in a ratio of 6:1. M6G is believed to be responsible for some additional analgesic effects of morphine [35]. M3G, on the other hand, is believed to potentially lead to hyperalgesia [36], with increased pain, agitation, and myoclonus. Morphine is also metabolized in small amounts to codeine and hydromorphone. For instance, in one study, hydromorphone was present in 66 % of morphine consumers without aberrant drug behavior [37]; this usually occurs with doses higher than 100 mg/day.
Codeine
It is believed that the analgesic activity from codeine occurs from metabolism of codeine to morphine by CYP2D6. Because of the great heterogeneity in the CYP2D6 enzyme, with both fast metabolizers and slow metabolizers, codeine may not be an effective drug in all populations. Recently, the FDA has issued a Public Health Advisory [38] regarding a serious side effect in nursing infants whose mothers are apparent CYP2D6 ultrarapid metabolizers, who, while taking codeine, had rapid and higher levels of morphine in the breast milk, with subsequent potentially fatal neonate respiratory depression.
Although codeine is often referred to as a “weak” analgesic, in a cancer pain study comparing 25 mg of hydrocodone (a “strong” analgesic) to 150 mg of codeine (a “weak” analgesic), 58 % of the codeine patients obtained relief compared to 57 % of the hydrocodone patients [39].
Hydrocodone
Hydrocodone is similar in structure to codeine and is a weak mu receptor agonist, but the CYP2D6 enzyme demethylates it into hydromorphone, which has much stronger mu binding [16]. Like codeine, it has been proposed that hydrocodone is a prodrug. In other words, patients who are CYP2D6 deficient, or patients who are on CYP2D6 inhibitors, may not produce the hydromorphone metabolites and may have less than expected analgesia.
Oxycodone
Oxycodone has activity at multiple opiate receptors including the kappa receptor, which gives it a unique anti-sedative effect (“perky Percocet”). It undergoes extensive hepatic metabolism by glucuronidation to noroxycodone (which has less than 1 % of the analgesia potency of oxycodone) and by CYP2D6 to oxymorphone [40]. Because oxycodone is dependent on the CYP2D6 pathway for clearance, it is possible that drug–drug interactions can occur with 2D6 inhibitors.
Oxymorphone
Although oxycodone has activity at multiple receptors, its metabolite oxymorphone is a pure mu agonist. Oxymorphone is about ten times more potent than morphine. It has limited protein binding and is not affected by CYP2D6 or CYP3A4, which decreases the risk of drug–drug interactions [41]. Oxymorphone has a reduced histamine effect and may be of use in patients who complain of headache or itching with other opioids [42].
Hydromorphone
Hydromorphone is a hydrogenated ketone of morphine [43]. Like morphine, it acts primarily on mu opioid receptors and to a lesser degree on delta receptors. While hydromorphone is 7–10 times more potent than morphine in single-dose studies [44], the oral and parenteral steady-state equivalence is 1:5, while the equivalence of chronic infusions may be as little as 1:3.5 [45]. It is highly water-soluble, which allows for very concentrated formulations, and in patients with renal failure, it may be preferred over morphine. Hydromorphone is metabolized primarily to hydromorphone-3-glucuronide (H3G), which, similar to the corresponding M3G, is not only devoid of analgesic activity but also evokes a range of dose-dependent excited behaviors including allodynia, myoclonus, and seizures in animal models [46].
Methadone
Methadone is a synthetic mu opioid receptor agonist medication. It is a racemic mixture of two enantiomers; the R form is more potent, with a tenfold higher affinity for opioid receptors (which accounts for virtually all of its analgesic effect), while S-methadone is the NMDA antagonist. The inherent NMDA antagonistic effects make it potentially useful in severe neuropathic and “opioid-resistant” pain states. The S isomer also inhibits reuptake of serotonin and norepinephrine, which should be recognized when using methadone in combination with SSRIs and TCAs. Although it has traditionally been used to treat heroin addicts, its flexibility in dosing, use in neuropathic pain, and cheap price have led to a recent increase in its use. Unfortunately, a lack of awareness of its metabolism and potential drug interactions, as well as its long half-life, has led to a dramatic increase in the deaths associated with this medication.
Methadone is unrelated to standard opioids, leading to its usefulness in patients with “true” morphine allergies. Methadone is metabolized in the liver and intestines and excreted almost exclusively in feces, an advantage in patients with renal insufficiency or failure.
The metabolism of methadone is always variable. Methadone is metabolized by CYP3A4 primarily and CYP2D6 secondarily; CYP2D6 preferentially metabolizes the R-methadone, while CYP3A4 and CYP1A2 metabolize both enantiomers. CYP1B2 is possibly involved, and a newly proposed enzyme CYP2B6 may be emerging as an important intermediary metabolic transformation [47]. CYP3A4 expression can vary up to 30-fold, and there can be genetic polymorphism of CYP2D6, ranging from poor to rapid metabolism. The initiation of methadone therapy can induce the CYP3A4 enzyme for 5–7 days, leading to low blood levels initially, but unexpectedly high levels may follow about a week later if the medication has been rapidly titrated upward. A wide variety of substances can also induce or inhibit these enzymes [48]. The potential differences in enzymatic metabolic conversion of methadone may explain the inconsistency of observed half-life.
Methadone has no active metabolites and therefore may result in less hyperalgesia, myoclonus, and neurotoxicity than morphine. It may be unique in its lack of profound euphoria, but its analgesic action (4–8 h) is significantly shorter than its elimination half-life (up to 150 h), and patient self-directed redosing and a long half-life may lead to the potential of respiratory depression and death.
Methadone also has the potential to cause cardiac arrhythmias, specifically prolonged QTc intervals and/or torsade de pointes under certain circumstances. Congenital QT prolongation, high methadone levels (usually over 60 mg/day), and conditions that increase QT prolongation (such as hypokalemia and hypomagnesemia) or IV methadone, because it contains chlorobutanol, which prolongs QTc intervals [49], may increase that risk [50]. Combining methadone with a CYP3A4 inhibitor such as ciprofloxin [51] potentially can increase that risk. It is recommended that a switch to methadone from another opioid be accompanied by a large (50–90 %) decrease in the calculated equipotent dose (Table 10.1) [53]. It cannot be too strongly emphasized that the dosing of methadone can be potentially lethal and must be done with knowledge and caution.
Table 10.1
Oral morphine to methadone conversion
Oral morphine dose (mg) | MS: methadone ratio |
|---|---|
30–90 | 4:1 |
90–300 | 8:1 |
300–800 | 12:1 |
800–1,000 | 15:1 |
>1,000 | 20:1 |
Fentanyl
Fentanyl is approximately 80 times more potent than morphine, is highly lipophilic, and binds strongly to plasma proteins. Fentanyl undergoes extensive metabolism in the liver. Fentanyl is metabolized by CYP3A4 to inactive and nontoxic metabolites [54]; however, CYP3A4 inhibitors may lead to increased fentanyl blood levels. The transdermal formulation has a onset of action lag time of 6–12 h after application and typically reaches steady state in 3–6 days. When a patch is removed, a subcutaneous reservoir remains, and drug clearance may take up to 24 h.
Conversion Tables
The usual recommendation for calculating the equipotent dose of different opioids involves calculating the 24-h dose as “morphine equivalents” (see Table 10.2). However, Hanks and Fallon [54] instead suggest relating the starting doses to 4-h doses of morphine rather than 24-h doses. For example, in patients receiving 5–20 mg oral morphine every 4 h (or the equivalent in controlled-release morphine), start with 25 mcg/h fentanyl patches every 72 h; patients on 25–35 mg oral morphine every 4 h, 50 mcg/h of fentanyl; patients on 40–50 mg oral morphine every 4 h, 75 mcg/h fentanyl; and patients on 55–65 mg oral morphine every 4 h, 100 mcg/h fentanyl. They feel that the controversies over appropriate morphine to fentanyl potency ratio calculations miss the point that fentanyl transdermally behaves differently and cannot be equated with oral routes when calculating relative potency.
Table 10.2
Opioid conversions
Drug | Initial po dose | PO:IV | PO MS:PO drug | PO drug: PO MS |
|---|---|---|---|---|
Morphine | 2.5–15 mg | 3:1 | 1:1 | 1:1 |
Hydromorphone | 1, 2, or 4 mg | 4:1 | 1:0.25 | 1:4 |
Oxycodone | 5 or 10 mg | N/A | 1:0.66 | 1:1.5 |
Oxymorphone | 2.5, 5, or 10 mg | 10:1 | 1:0.33 | 1:3 |
Methadone | 2.5 or 5 mg | 2:1 | a | a |
TD fentanyl | 25 mcg/hb | TD = IV/h | b | b |
Tramadol
A unique analgesic, tramadol, is an atypical synthetic analogue of codeine [56]. The M1 derivative (O-demethyl tramadol) produced by CYP2D6 has a higher affinity for the mu receptor than the parent compound (as much as six times). Tramadol is a racemic mixture of two enantiomers—one form is a selective mu agonist and inhibits serotonin reuptake, while the other mainly inhibits norepinephrine reuptake [57]. Maximum dose is 400 mg/day, and toxic doses cause CNS excitation and seizures. Because it requires CYP2D6 metabolism for maximal analgesic effect, coadministration of CYP2D6 inhibitors such as fluoxetine, paroxetine, and sertraline is contraindicated. In addition, because tramadol has serotonin activity, SSRIs are relatively contraindicated because of the potential of a serotonin syndrome.
Although considered a “weak” opioid, it can have significant analgesic qualities (perhaps because of its dual opioid and SNRI action). In a study of 118 patients with moderate to severe cancer pain comparing 25 mg of hydrocodone to 200 mg of tramadol, 62 % of the tramadol patients obtained relief, compared to 57 % of the hydrocodone patients [58].
Opioid Routes of Administration
Oral
Major advances in the pharmacotherapy of chronic pain have led to the development of extended-release opioid delivery systems, thereby allowing less frequent dosing than the classic short-acting formulas. It is the patterns in serum drug levels that define the difference between short-acting opioids (SAO) and long-acting opioids (LAO); with SAOs, serum opioid levels rise rapidly following administration and then decline rapidly, while LAO administration allows for less fluctuation in serum opioid levels and an extended period within the therapeutic range (Fig. 10.4) [59]. The assumption that plasma levels of opioids correspond to analgesia has led to the additional concept of minimum effective concentration (MEC), the plasma level of an opioid below which there is ineffective analgesia.
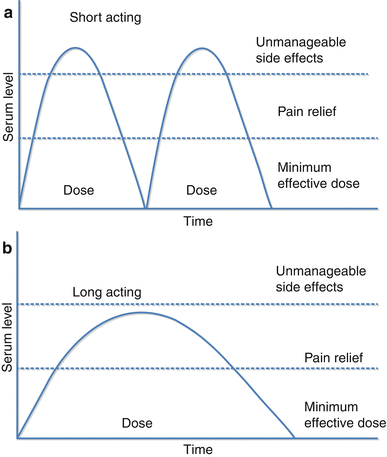
Fig. 10.4
Serum drug level following administration of (a) short-acting and (b) long-acting opioids (Modified from McCarberg and Barkin [59])
There are many proposed advantages of the long-acting opioid formulas compared to the short-acting formulas. Because of the longer duration of action, there is a lessening of the frequency and severity of end-of-dose pain [60]. Furthermore, it has been suggested that less frequent dosing leads to increased compliance and improved efficacy [61]. Sustained analgesia and uninterrupted sleep are other potential advantages of the extended-release formulation compared to the short-acting variety. However, in a recent systematic review of long-acting versus short-acting opioids, Rauck [62] noted that, while it was clear that long-acting opioids achieved more stable drug levels, there was no clear evidence from appropriately designed comparative trials to make a case for the use of one type of formulation over the other on the basis of clinical efficacy.
Transmucosal
Oral transmucosal fentanyl citrate (OTFC) has become a mainstay in the treatment of breakthrough pain because it provides faster absorption of the lipophilic fentanyl than any other oral opioid formulation [63]. This “fentanyl lollipop” consists of medication on the end of a stick, which is applied to the buccal membrane. A newer formulation of fentanyl, the fentanyl buccal tablet (FBT), was designed to provide an even faster relief. Additional delivery systems for intranasal and inhaled fentanyl are being developed [64].
Intravenous
Intravenous delivery of opioids allows for rapid and reliable delivery of medicine, but veins for administration are not always available. In general, the IV dose is approximately 1/3 of the oral dose, since IV medications do not have a first-pass effect. Opioids can be delivered intermittently or continuously; patient-controlled analgesia (PCA) is now available for outpatient use so that small doses of opioids are delivered when the patient pushes a button, with or without a continuous infusion of opioid.
Subcutaneous
Subcutaneous opioid injections can be an option for the patient unable to tolerate oral medications but without IV access. The medication is administered through a butterfly needle and can be given intermittently or continuously. Onset is slower and lower peak effect than IV, but this may be a better option for acute or escalating pain than transdermal fentanyl, which has an even slower onset and prolonged effect [65]. Subcutaneous infusions up to 10 cc/h can be usually absorbed, but patients are usually more comfortable with 2–3 cc/h.
Rectal
The rectal mucosa absorbs many medications easily, including most opioids, and the blood flow from the rectum bypasses the liver so that rectal morphine results in blood levels that are almost 90 % of the oral dose [66]. A double-blind, double-dummy, crossover study in 1995 compared oral versus rectal morphine, which was shown to be effective, easy to manage, and inexpensive, with a rapid onset of action [67].
Related posts:
 The Role of Antidepressants in the Treatment of Chronic Pain
Anticonvulsant Medications for Treatment of Neuropathic and “Functional” Pain
Polypharmacy and Drug Interaction
The Future of Pain Pharmacotherapy
The Role of Antidepressants in the Treatment of Chronic Pain
Anticonvulsant Medications for Treatment of Neuropathic and “Functional” Pain
Polypharmacy and Drug Interaction
The Future of Pain Pharmacotherapy
 Opioid Adverse Effects and Opioid-Induced Hypogonadism
Opioid Adverse Effects and Opioid-Induced Hypogonadism
 Acute Management of the Opioid-Dependent Patient
Acute Management of the Opioid-Dependent Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


